Machine learning approaches are increasingly being used to aid small-molecule structure elucidation from mass spectrometry data.
Within metabolomics, one area positioned to benefit from artificial intelligence (AI) is molecular structure elucidation for small molecules from liquid chromatography–tandem mass spectrometry (LC–MS/MS).
Recognizing this potential, the community has devoted extensive effort to developing datasets and ML models for automated structure elucidation2,3,4,5,6,7,8,9,10.
However, past studies report that ML methods for structure elucidation fall short on this task7,11.
Formulating mass spectrometry analysis as machine translation enables the use of powerful architectures developed for machine translation, especially the transformers network15.
Machine learning approaches are increasingly being used to aid small-molecule structure elucidation from mass spectrometry data. Surprisingly, however, current models often fail to outperform even simple baseline methods. Here we examine why these approaches fall short and propose strategies to overcome their limitations.
The advent of machine learning (ML) has led to transformative breakthroughs in biology. For example, the development of AlphaFold1 has significantly advanced protein structure prediction and accelerated drug discovery. Within metabolomics, one area positioned to benefit from artificial intelligence (AI) is molecular structure elucidation for small molecules from liquid chromatography–tandem mass spectrometry (LC–MS/MS). Because manual spectral interpretation is time-consuming and labour-intensive, automating this process would enable high-throughput compound identification at scale and fundamentally reshape the field.
Recognizing this potential, the community has devoted extensive effort to developing datasets and ML models for automated structure elucidation2,3,4,5,6,7,8,9,10. However, past studies report that ML methods for structure elucidation fall short on this task7,11. Given continual advances in general AI methodology and access to large spectral datasets, these findings are puzzling. As such, advancing structure elucidation requires understanding the failure modes of current technologies. We begin by summarizing how current methods work and evaluating their performance.
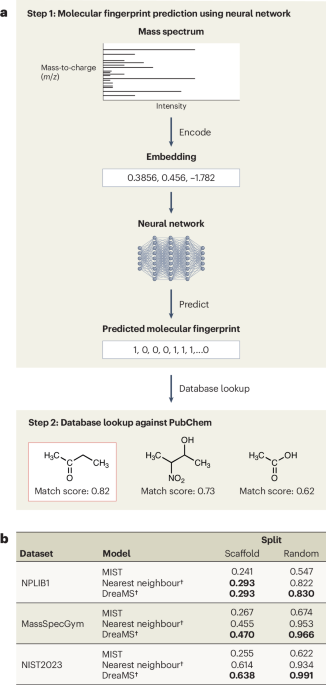
Most current AI approaches for this task share a common two-step pipeline (Fig. 1a). Given an experimental spectrum, an ML model predicts a molecular fingerprint, which is then used to query molecular databases such as PubChem12 to retrieve candidate molecules. This approach can potentially identify compounds that have never been previously characterized by mass spectrometry, as it can in principle map any spectrum to a molecular fingerprint. This workflow is a major departure from traditional pre-AI methods, which rely on spectral library matching and can only identify molecules whose spectra already exist and are labelled in reference databases. From an algorithmic perspective, this mapping approach is analogous to machine translation in natural language processing (NLP), whereby a source sentence is encoded into a vector (also known as embedding) and then decoded into its translation in the target language. In this analogy, spectral peaks serve as ‘words’, collectively forming a ‘sentence’ that is ‘translated’ into a molecular fingerprint. As in ML translation, the ML algorithm is trained on a dataset of experimental spectra paired with molecular fingerprints. Commonly used datasets include NPLIB1 (ref. 13), MassSpecGym3 and the NIST 2023 LC–MS/MS dataset14. Fig. 1: Pipeline for chemical structure elucidation using ML models. The alternative text for this image may have been generated using AI. Full size image a, Step 1: Given an LC–MS/MS spectrum, the model first encodes it into a vector embedding. This embedding is then used by a neural network to predict the molecular fingerprint of the molecule that generated the spectrum. Step 2: The predicted fingerprint is compared against the fingerprints of known molecules in public databases such as PubChem. Candidate molecules are ranked on the basis of their similarity to the predicted fingerprint. The structure of the molecule with the highest similarity score is then assigned as the label for the spectrum. b, Matching performance measured by the binary Jaccard score using Morgan fingerprints (4,096 bits, radius = 2) for all models under different dataset splitting strategies. † indicates zero-shot evaluation (no task-specific training). For the nearest-neighbour and DreaMS baselines, predictions are obtained via top-1 retrieval of the most similar training example. Bolded records indicate the best-performing model.
Formulating mass spectrometry analysis as machine translation enables the use of powerful architectures developed for machine translation, especially the transformers network15. Their strength lies in building a vector space for representing text meaning: they encode words and sentences into embeddings such that semantically similar inputs are geometrically close. The same mechanism enables generation of molecular fingerprints from spectral embeddings. Given the strong performance of transformer networks across many domains, framing spectrum-to-fingerprint prediction as a translation task holds significant promise for the LC–MS/MS setting.
However, treating peaks and spectra naively as words and sentences is suboptimal. While words may have several meanings, the ambiguity of fragment mass peaks is significantly higher as a single peak can correspond to a much larger number of possible substructures. Moreover, spectra are inherently noisy. Some peaks may correspond to other components of the molecular mixture, without contributing useful structural information to the target molecule — unlike words, which generally contribute to sentence meaning.
To address these issues, ML approaches preprocess spectra into representations more amenable to the translation paradigm. Goldman et al.5 map m/z values to candidate chemical formulas, making peaks more word-like. Bushuiev et al.4 encode peaks using learnable Fourier features, enabling the model to capture higher-order relationships between masses — analogous to learnt word relationships in NLP. Another concept adopted from NLP is foundation models, which learn input representations from large amounts of raw data using auxiliary objectives (for example, masking). Bushuiev et al.4 leverage this strategy to learn spectral embeddings, which are then fine-tuned using paired spectra–fingerprint data. A detailed explanation of key ML terms used in this Comment is provided in Supplementary Table 1.
As with all ML systems, the key question is whether models generalize to unseen inputs, measuring their capacity to analyse molecules different from the ones seen in training. How can we evaluate generalization? The easiest evaluation relies on random splits, in which spectra are divided into non-overlapping training and test sets. While this approach controls for spectral overlap, the same molecule may appear in both sets as different spectra, resulting in data leakage. A more informative and practically useful evaluation uses scaffold splits, in which molecules are separated by chemical scaffolds. Another way to assess generalization and the practical utility of ML models is by comparing their performance against non-ML approaches used today. A commonly used method is a nearest-neighbour baseline, in which a test spectrum is assigned the molecular fingerprint of its closest training counterpart according to the cosine similarity metric. The performance of all ML approaches is evaluated using the binary Jaccard score between predicted and ground-truth molecular fingerprints.
Figure 1b shows the performance of two state-of-the-art ML methods: MIST5, a fingerprint-prediction model, and DreaMS4, a retrieval-based foundation model, together with the nearest-neighbour retrieval baseline, across three benchmark datasets. A detailed description of the task formulation, evaluation metrics and benchmarked models is provided in Supplementary Notes 1–3, while data attribution methods and additional analyses are described in Supplementary Notes 4 and 5.
The results are surprising. The performance across all ML methods is poor. On the scaffold split, nearest-neighbour outperforms MIST and nears the performance of DreaMS. DreaMS, the top model under the random split, drops sharply under the scaffold split, showing poor generalization. Even more puzzling is the modest performance on an easy setting (the random split) where there is high molecular overlap between training and test sets (51.9% for NPLIB1, 92.3% for MassSpecGym and 99.5% for NIST2023). These findings contradict the hypothesis that low performance arises primarily from insufficient training coverage11. Simply adding more data to existing models is unlikely to solve the problem.
To understand these failures, we turn to data-attribution methods, which trace model performance back to the data from which the algorithm learns. These methods identify examples that are hard for the model to reason about, leading to the ultimate performance drop. We use two complementary approaches — influence functions and learning-to-split — to ensure robustness. Influence functions identify training examples that help or hurt specific predictions. Learning-to-split instead takes all the data and divides them into training and test sets, so the test performance is the lowest. De facto, it selects test examples that are the hardest for the algorithm to understand. By analysing these examples, we can get a better understanding of how the algorithms fail. Below we summarize results of these analyses applied to ML algorithms for LC–MS/MS.
Inability to generalize across experimental conditions Both learning-to-split and influence functions flag spectra of molecules collected under different conditions as challenging. While current datasets contain data from multiple experimental settings, algorithms fail to model how change in conditions impacts the spectra.
Inability to capture peak intensity Data attribution methods show that models struggle to distinguish molecules with similar m/z distributions but different intensity profiles. In fact, regardless of intensity, the algorithm maps spectra with similar m/z patterns into similar vector representations, largely ignoring intensity information.