
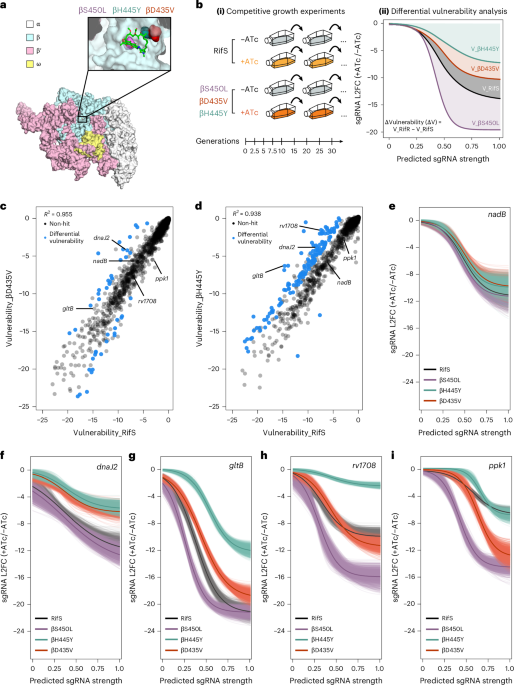
Transcription attenuation amplifies collateral vulnerabilities in rifampicin-resistant Mycobacterium tuberculosis
By keeping the OD 600 of the 20 ml cultures ≥0.05, we guaranteed sufficient coverage of the library (3,000×) at all times. Cultures were grown for 3 days and then expanded to 2 × 7.5 ml cultures. Double-stranded DNA was synthesized using DNA Polymerase I (New England BioLabs, M0209S). Mtb cultures were grown to a late-logarithmic phase (OD 600 ≈ 0.8) and then back diluted to a starting OD 600 of 0.01. Mtb cultures were grown to a late-logarithmic phase (OD 600 ≈ 0.8) and then back diluted to a starting OD 600 of 0.01.
Bacterial strains
Mtb strains are derivatives of H37Rv unless otherwise noted. ΔbioA Mtb was obtained from the Dirk Schnappinger laboratory26. E. coli strains are derivatives of DH5α (NEB). Strains are available upon reasonable request to the corresponding author.
Mycobacterial cultures
Mtb were grown at 37 °C in Difco Middlebrook 7H9 broth or on 7H10 agar supplemented with 0.2% glycerol (7H9) or 0.5% glycerol (7H10), 0.05% Tween-80, 1× oleic acid-albumin-dextrose-catalase (OADC, Mtb) and the appropriate antibiotics (kanamycin 10–20 μg ml−1 and/or hygromycin 25–50 μg ml−1 and/or zeocin 5–20 μg ml−1). ATc was used at 100 ng ml−1. Note that both 7H9 and 7H10 media are normally supplemented with biotin (0.5 mg l−1, ~2 μM), thereby allowing growth of the ΔbioA Mtb auxotroph. Mtb cultures were grown standing in tissue culture flasks (unless otherwise indicated) with 5% CO 2 . Relative growth of individual CRISPRi strains was determined by spotting assay. Tenfold serial dilutions (starting at 50,000 cells per spot) were plated on 7H10 with or without 100 ng ml−1 ATc. Plates were incubated at 37 °C and imaged after 14 days.
Selection of Rif-resistant isolates
For the selection of RifR H37Rv and ΔbioA, 5 independent 5-ml cultures were started at a density of ~2,000 cells per ml (to minimize the probability of seeding cultures with pre-existing RifR bacilli) and grown to stationary phase (optical density at 600 nm (OD 600 ) > 1.5). Cultures were pelleted (~3,900 × g for 10 min), resuspended in 30 μl remaining medium per pellet and plated on 7H10 agar supplemented with Rif at 0.5 μg ml−1. After outgrowth, colonies were picked into 7H9 medium. After 1 week of outgrowth, an aliquot was heat inactivated and the Rif-resistance-determining region of rpoB, rpoA and rpoC were amplified by PCR and Sanger sequenced (see Supplementary Table 5).
Generation of structural models
The structural model of Mtb RNAP transcription initiation complex bound to Rif was generated by modelling Mycobacterium smegmatis RNAP bound to Rif (PDB 6CCV)59 onto the transcription initiation complex structure (PDB 6EDT)60.
Generation of individual CRISPRi strains
plRL58 (Addgene, 166886; Supplementary Table 5) contains (1) the Sth1 dcas9 allele under the control of an optimized, synthetic Tet repressor (TetR)-regulated promoter; (2) the Sth1 sgRNA under the control of a synthetic TetR-regulated promoter; (3) a mycobacterial codon optimized Tet repressor; (4) a single-copy L5-integrating backbone61, with the integrase removed to increase plasmid stability; (5) a pBR322-derived E. coli replication origin; and (6) a kanamycin-selectable marker. To integrate plRL58 into the mycobacterial chromosome, L5 integrase function is supplied in trans on a separate suicide vector, plRL19 (Addgene, 163634; Supplementary Table 5).
Individual CRISPRi plasmids were cloned by restriction ligation cloning62. In brief, plRL58 was digested with BsmBI-v2 (NEB, R0739L) and gel purified. sgRNAs were designed to target the non-template strand of the target gene ORF. For each individual sgRNA, two complementary oligonucleotides with appropriate sticky end overhangs were annealed and ligated (T4 ligase, NEB, M0202M) into the BsmBI-digested plasmid backbone. Successful cloning was confirmed by Sanger sequencing. Individual CRISPRi plasmids were then electroporated into the appropriate mycobacterial species. Electrocompetent cells were obtained as previously described63. In brief, a bacterial culture was expanded to OD 600 = 0.4–0.6 and treated with glycine (final concentration 0.2 M) for ~1 doubling before pelleting ( ~3,900 x g for 10 min). The cell pellet was washed three times in sterile 10% glycerol. The washed bacilli were then resuspended in 10% glycerol in a final volume of 5% of the original culture volume. For each transformation, 100 ng plasmid DNA and 50–100 μl electrocompetent mycobacteria were mixed and transferred to a 2 mm electroporation cuvette (Bio-Rad, 1652082). Where necessary, 100 ng plasmid plRL19 (Addgene, plasmid 163634) was also added. Electroporation was performed using the Gene Pulser X cell electroporation system (Bio-Rad, 1652660) set at 2,500 V, 700 Ω and 25 μF. Electroporated cells were immediately recovered in 1 ml of complete 7H9 and cultured at 37 °C for 1 doubling, after which cells were pelleted at ~1,800 × g for 10 min, resuspended in 100 μl of 7H9 and plated on selective 7H10 plates. Colonies were picked after 14–21 days.
CRISPRi library transformation
Fifty transformations were performed to generate RifS, βS450L, βD435V and βH445Y ΔbioA libraries. For each transformation, 1 μg of RLC12 plasmid DNA was added to 100 μl (ΔbioA) electrocompetent cells. The cells:DNA mix was transferred to a 2 mm electroporation cuvette (Bio-Rad, 1652082) and electroporated at 2,500 kV, 700 Ω and 25 μF. Each transformation was recovered in 2 ml 7H9 medium supplemented with OADC, glycerol and Tween-80 (100 ml total) for 16–24 h. The recovered cells were collected at ~1,800× g for 10 min, resuspended in 400 μl remaining medium per transformation and plated on 7H10 agar supplemented with kanamycin (see ‘Mycobacterial cultures’) in Corning Bioassay dishes (Sigma, CLS431111-16EA). Note that the βH445Y library was generated and passaged in the presence of 0.05 μg ml−1 Rif (2.5× RifS MIC) to select against bacilli that reverted to a WT rpoB allele.
After 21 days of outgrowth on plates, transformants were scraped into 25% glycerol and pooled. Scraped cells were homogenized by two dissociation cycles on a gentleMACS Octo Dissociator (Miltenyi Biotec, 130095937) using the RNA_01 programme and 30 gentleMACS M tubes (Miltenyi Biotec, 130093236). The Mtb libraries were further declumped by passaging 1–3 ml of homogenized library into 100 ml of 7H9 supplemented with kanamycin (see ‘Mycobacterial cultures’) for between 2.5 and 10 generations. Final Mtb library stocks were obtained after passing the cultures through a 10-μm cell strainer (Pluriselect SKU 43-50010-03). Genomic DNA was extracted from the final stocks and library quality was validated by deep sequencing24 (see ‘Genomic DNA extraction and library preparation for Illumina sequencing of CRISPRi libraries’).
Pooled CRISPRi screen
Cultures (20 ml) were grown in vented tissue culture flasks (T-75, Falcon, 353136). 7H9 medium was supplemented with kanamycin (see ‘Mycobacterial cultures’) and maintained at 37 °C and 5% CO 2 in a humidified incubator. The screen was initiated by thawing 4× 1-ml aliquots of the Mtb ΔbioA (RifS or RifR mutant) CRISPRi library (RLC12) and inoculating each aliquot into 24 ml 7H9 medium supplemented with kanamycin in a T-75 flask (starting OD 600 ∼0.06). The cultures were expanded to OD 600 ≈ 1.0, pooled and passed through a 10-μm cell strainer (pluriSelect, 43-50010-03) to obtain a single-cell suspension. The single-cell suspension (flow-though) was used to set up 6 ‘generation 0’ cultures: 3 replicate cultures with ATc (+ATc) and 3 replicate control cultures without ATc (−ATc). From each generation 0 culture, we collected 10 OD 600 units of bacteria (∼3 × 109 bacteria; ∼30,000× coverage of the CRISPRi library) for genomic DNA extraction. The remaining culture volume was used to initiate the pooled CRISPRi fitness screen. Cultures were periodically passaged in pre-warmed medium to maintain log-phase growth. At generation 2.5, 5 and 7.5, cultures were back diluted 1:6 (to a starting OD 600 = 0.2) and cultivated for ~2.5 doublings. At generation 10, 15, 20 and 25, cultures were back diluted 1:24 (to a starting OD 600 = 0.05) and expanded for 5 generations before reaching late-log phase. ATc was replenished at every passage. By keeping the OD 600 of the 20 ml cultures ≥0.05, we guaranteed sufficient coverage of the library (3,000×) at all times. At defined timepoints (~2.5, 5, 7.5, 10, 15, 20, 25 and 30 generations), we collected bacterial pellets (10 OD 600 units) to extract genomic DNA24. The βH445Y library was generated and passaged in the presence of 0.05 μg ml−1 rifampicin (2.5× the RifS MIC) to prevent reversion to a wild-type rpoB allele. This rifampicin concentration had no measurable impact on the growth of βH445Y. To confirm genetic stability during passaging, whole-genome sequencing was performed on one +ATc and one −ATc replicate at 3 timepoints (0, 15 and 30 generations) for the βS450L, βD435V and βH445Y libraries. These analyses confirmed that the RifR libraries neither reverted to RifS nor acquired compensatory mutations in rpoA, rpoC, rpoB or nusG.
Pooled CRISPRi chemical-genetic screening
Chlorflavonin chemical-genetic screening was performed as described in ref. 34. The chemical-genetic screen was initiated by thawing a 1-ml aliquot of the above ΔbioA (RifS or βS450L) CRISPRi library (RLC12; Addgene, 163954) and inoculating into 5 ml 7H9 supplemented (approximate starting OD of 0.2) with kanamycin (10 μg ml−1) in a vented tissue culture flask (T-25, Falcon, 353109). Cultures were grown for 3 days and then expanded to 2 × 7.5 ml cultures. These cultures were grown to OD 600 ~1.0, pooled and passed through a 10-μm cell strainer (pluriSelect, 43-50010-03) to obtain a single-cell suspension. The single-cell suspension was then treated with ATc (100 ng ml−1 final concentration) to initiate target pre-depletion. To generate a 5-day pre-depletion culture, the culture was diluted to a starting OD 600 of 0.1 in 40 ml with 100 ng ml−1 ATc. To initiate the chemical-genetic screen, we first collected 10 OD 600 units of bacteria (~3 × 109 bacteria; ~30,000× coverage of the CRISPRi library) from the 5 day CRISPRi library pre-depletion cultures as input controls. Triplicate cultures were then inoculated at OD 600 = 0.05 in 10 ml 7H9 supplemented with ATc (100 ng ml−1), kanamycin (10 μg ml−1) and the indicated drug concentration (Extended Data Fig. 3a) or dimethyl sulfoxide (DMSO) vehicle control. Pooled CRISPRi chemical-genetic screens were performed in vented tissue culture flasks (T-25, Falcon, 353109). Cultures were outgrown for 14 days at 37 °C and 5% CO 2 . ATc was replenished at 100 ng ml−1 at day 7. After 14 days outgrowth, OD 600 values were measured for all cultures to empirically determine the MIC for each drug. Samples from two descending doses of partially inhibitory drug concentrations were processed for genomic DNA extraction and for next generation sequencing (NGS). Concentrations were chosen to best match the degree of growth inhibition compared to the vehicle control for RifS and βS450L libraries: RifS high = 0.85 μg ml−1 (75.6 ± 4.4% growth), low = 0.425 μg ml−1 (97.8 ± 4.7% growth), βS450L high = 1.1 μg ml−1 (57.1 ± 3.1% growth), low = 0.55 μg ml−1 (107.5 ± 7.7% growth).
Genomic DNA extraction and library preparation for Illumina sequencing of CRISPRi libraries
Genomic DNA was isolated from bacterial pellets using the CTAB–lysozyme method described previously64, or using a modified version of the mechanical lysis method described in ref. 65. Briefly, bacterial pellets were resuspended in 600 μl TE buffer (10 mM Tris, 1 mM EDTA pH 8.0) in 2 ml tubes containing lysing matrix B (MP Biomedicals, 116911050). One volume of phenol–chloroform–isoamyl alcohol (PCI) (25:24:1) was added and samples were lysed with the Precellys Evolution homogenizer (Bertin, 02520-300-RD000) at 10,000 r.p.m. for 2 × 30 s with 5 min chilling on a freezer block or ice. After removal of the beads by spinning samples at ~12,000 × g for 10 min at 4 °C, the aqueous phase was transferred to a new tube containing 50 μl 5 M NaCl. After mixing by inversion, one volume of PCI was added, and samples were incubated at room temperature for ~30 min. Samples were spun at ~12,000 × g for 10 min at 4 °C, and the aqueous phase was transferred to a new tube. RNAse A (3 μl; Thermo Scientific, EN0531) was added and samples were incubated at 37 °C for 45 min. One volume of chloroform was added, followed by mixing with vigorous shaking. Samples were allowed to sit for 10 min. Samples were spun at ~12,000 × g for 10 min at 4 °C, and the aqueous phase was transferred to a new tube with 1/10 volume 3 M sodium acetate pH 5.2 (NaOAc). One volume of cold isopropanol was added, and samples were incubated at −20 °C for 1–4 days before spinning at ~12,000 × g for 45 min at 4 °C. Genomic DNA was washed 2× with freshly prepared cold 70% ethanol before resuspending in 50 μl DNAse and RNAse-free water. Genomic DNA concentration was quantified using the DeNovix dsDNA high sensitivity assay (KIT-DSDNA-HIGH-2, DS-11 Series spectrophotometer/fluorometer).
To construct Illumina libraries, the sgRNA-encoding region was amplified from 500 ng genomic DNA using NEBNext Ultra II Q5 master Mix (NEB M0544L). PCR cycling conditions were: 98 °C for 45 s; 17 cycles of 98 °C for 10 s, 64 °C for 30 s, 65 °C for 20 s; 65 °C for 5 min. Each PCR reaction contained a unique indexed forward primer (0.5 μM final concentration) and a unique indexed reverse primer (0.5 μM) (Supplementary Table 5). Forward primers contained a P5 flow-cell attachment sequence, a standard Read1 Illumina sequencing primer binding site, custom stagger sequences to ensure base diversity during Illumina sequencing, and unique barcodes to allow for sample pooling during deep sequencing. Reverse primers contained a P7 flow-cell attachment sequence, a standard Read2 Illumina sequencing primer binding site and unique barcodes. Following PCR amplification, each ∼230 bp amplicon was purified using AMPure XP beads (Beckman Coulter, A63882) using two-sided selection (0.75× and 0.12×). Eluted amplicons were quantified with a Qubit 2.0 Fluorometer (Invitrogen), and amplicon size and purity were quality controlled by visualization on an Agilent 4200 Tape Station (Instrument: Agilent, G2991AA; reagents: Agilent, 5067-5583; tape: Agilent, 5067-5582). Next, individual PCR amplicons were multiplexed into 20–50 nM pools and sequenced on an Illumina sequencer according to manufacturer instructions. To increase sequencing diversity, a PhiX spike-in of 2.5–10% was added to the pools (PhiX sequencing control v3; Illumina, FC-110-3001). Samples were run on the Illumina NextSeq 500, NovaSeq 6000 and NovaSeqX Plus platforms (single-read 1 × 85 cycles, 8 × i5 index cycles and 8 × i7 index cycles)24,34.
Differential vulnerability analysis of Rif-resistant versus Rif-sensitive strains
Gene vulnerability in the RifS and RifR M. tuberculosis strains was determined using an updated version of our previously described vulnerability model24. In the updated model, read counts for a given sgRNA in the −ATc conditions were modelled using a negative binomial distribution with a mean proportional to the counts in the +ATc condition, plus a factor representing the log 2 (fold change):
$${y}_{i}^{-\mathrm{ATc}} \sim \mathrm{NegBinom}\left({\eta }_{i},\phi \right)$$ (1)
$${\eta }_{i}=\log \left({y}_{i}^{+\mathrm{ATc}}+{\lambda }_{i}\right)+\mathrm{TwoLine}\left({x}_{i},{\alpha }_{l},{\beta }_{l},\gamma ,{\beta }_{e}\right)$$ (2)
where \(\,{\lambda }_{i}\) is an sgRNA-level correction factor estimated by the model, \({x}_{i}\) represents the generations analysed for the ith guide, and the TwoLine function represents the piecewise linear function previously described, which models sgRNA behaviour over time. The logistic function describing gene-level vulnerabilities was simplified by setting the top asymptote of the curve (previously ‘K’) equal to 0, representing the fact that weakest possible sgRNAs are expected to impose no effect on bacterial fitness, that is:
$$\mathrm{Logistic}\left(s\right)=\frac{{\beta }_{\max }}{\left(1+{e}^{\left(-H\cdot \left(s-M\right)\right)}\right)}$$ (3)
The Bayesian vulnerability model was run for each condition independently, and samples for all the parameters were obtained using Stan running 4 independent chains with 1,000 warmup iterations and 3,000 samples each (for a total of 12,000 posterior samples for each parameter in the model after discarding warmup iterations).
Differential vulnerabilities were estimated using two approaches. First, for each gene, the difference in pairwise (guide-level) vulnerability estimates was obtained, resulting in posterior samples of the differential vulnerability (delta vulnerability). This effectively estimated the difference in the integrals of the vulnerability functions. If the 95% credible region did not overlap 0, those were taken as significant differential vulnerabilities between the strains. Next, to identify differences between genes that may not exhibit the expected dose–response curve, we estimated the fitness cost (log 2 FC) predicted by our model for a (theoretical) sgRNA of strength 0 (that is, \(\mathrm{Logistic}\left(s=0\right)\)). This represented the weakest phenotype theoretically possible with our CRISPRi system, which we call ‘\({F}_{\min }\)’. The difference between these F min values was estimated for each gene (\(\Delta {F}_{\min }\)) and those where the 95% credible region did not overlap 0 were identified as significant differential vulnerabilities by this approach.
Single-stranded (ss)DNA recombineering and validation of strains
Attenuator disruption mutations were introduced into RifS and βS450L Mtb using oligo-mediated (ssDNA) recombineering as described in ref. 63. Briefly, 70-mer oligos were designed to correspond to the lagging strand of the replication fork, with the desired mutation in the middle of the sequence. Alterations were chosen to avoid recognition by the NucS mismatch-repair machinery. RecT expression was induced ~16 h before transformation by addition of ATc to a final concentration of 0.5 μg ml−1. Competent cells (400 μl) were transformed with 5 μg of mutation containing oligo and 0.1 μg of hygromycin resistance cassette repair oligo (1:50 ratio of mutant oligo to repair oligo) and recovered in 5 ml 7H9 media.
After 24 h of recovery, 200 μl of cells were plated on 7H10 plates supplemented with hygromycin. After 21 days of outgrowth, 12 colonies per construct were picked into 100 μl 7H9 media supplemented with hygromycin in a 96-well plate (Fisher Scientific, 877217). A volume of 50 μl of culture were heat inactivated at 80 °C for 2 h in a sealed microamp 96-well plate (Fisher Scientific, 07200684; Applied Biosystems, N8010560). A volume of 50 μl of heat-inactivated culture was mixed with 50 μl 25% DMSO and lysed at 98 °C for 10 min. Mutations of interest were confirmed by PCR amplification and Sanger sequencing. The region of interest was PCR amplified with NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541L) using 0.5 μl of heat-lysed product with the appropriate primers, annealing temperatures and extension times (see Supplementary Table 5). Amplicons were then submitted for Sanger sequencing.
RNA extraction and RT–qPCR
Approximately 2 OD 600 units of bacteria were added to an equivalent volume of GTC buffer (5 M guanidinium thiocyanate, 0.5% sodium N-lauroylsarcosine, 25 mM trisodium citrate dihydrate and 0.1 M 2-mercaptoethanol), pelleted by centrifugation, resuspended in 1 ml TRIzol (Thermo Fisher, 15596026) and lysed by zirconium bead beating (MP Biomedicals, 116911050). Chloroform (0.2 ml) was added to each sample and phases were separated by centrifugation. The aqueous phase was then purified using a Direct-zol RNA miniprep kit (Zymo, R2052). Residual genomic DNA was removed by TURBO DNase treatment (Invitrogen Ambion, AM2238). After RNA cleanup and concentration (Zymo, R1017), 3 μg RNA per sample was reverse transcribed into complementary (c)DNA with random hexamers (Thermo Fisher, 18–091-050) following manufacturer instructions. RNA was removed by alkaline hydrolysis, and cDNA was purified with PCR cleanup columns (Qiagen, 28115)24. Next, changes in target gene expression were quantified using TaqMan fluorescent dye-based quantitative real-time PCR (Thermo Fisher, 4444557) on a Quantstudio System 5 (Thermo Fisher, A28140) with gene-specific qPCR primers (10 μM) and probe (10 μM), normalized to sigA (rv2703) and quantified by the ΔΔC t algorithm. All gene-specific qPCR primers and probes were designed using the PrimerQuest tool from IDT (https://www.idtdna.com/PrimerQuest/Home/Index) and then validated for efficiency and linear range of amplification using standard qPCR approaches. Specificity was confirmed for each validated qPCR primer pair through melting curve analysis. All qPCR primers used in this study can be found in Supplementary Table 5.
SEnd-seq
Total RNA extraction was performed as described above, without the use of RNA miniprep or clean and concentrator columns to preserve low abundance and short RNA transcripts. Instead, after chloroform addition, the aqueous phase was mixed 1:1 with 100% isopropanol and placed at −20 °C for 2 h, then centrifuged at ~12,000 × g for 15 min at 4 °C. The pellet was washed twice with 1 ml 75% (v/v) ethanol, air dried for 5 min and dissolved in nuclease-free water36. Subsequently, the samples were treated with 0.5 µl TURBO DNase (Invitrogen Ambion, AM2238) at 37 °C for 15 min. The RNA was then purified twice using chloroform:isoamyl alcohol (Thermo Fisher, 15593031) and recovered by ethanol precipitation.
To construct SEnd-seq libraries, 5 µg of total RNA was mixed with pooled spike-in RNAs at a mass ratio of 300:1 in a total volume of 12 μl66. The RNA sample was incubated with a 5′-adaptor ligation mix (1 μl 100 μM 5′ adaptor (Supplementary Table 5), 0.5 μl 50 mM ATP, 2 μl DMSO, 5 μl 50% PEG8000, 1 μl RNase Inhibitor (New England BioLabs, M0314) and 1 μl High Concentration T4 RNA Ligase 1 (New England BioLabs, M0437)) at 23 °C for 5 h. The sample was then diluted to 40 μl with nuclease-free water and cleaned twice with 1.5× volume of Agencourt RNAClean XP beads (Beckman Coulter, A63987). Immediately following the 5′ adaptor ligation, the eluted RNA was ligated to the 3′ adaptor (Supplementary Table 5) using the same procedure. After incubation at 23 °C for 5 h, the reaction was diluted to 40 μl with water and cleaned twice with 1.5× volume of Agencourt RNAClean XP beads to remove excess adaptors. The sample was subsequently eluted with 0.1× TE buffer and subjected to ribosomal RNA removal with RiboMinus Transcriptome Isolation kit (Thermo Fisher, K155004) following manufacturer instructions. After recovery by ethanol precipitation, the RNA was reverse transcribed to cDNA with Eubacterium rectale maturase (recombinantly purified from Eco67, obtained from A. M. Pyle) and 5′-phosphorylated and biotinylated reverse transcription primer (Supplementary Table 5). After purification, the cDNA was circularized with TS2126 RNA ligase68 (obtained from K. Ryan). Double-stranded DNA was synthesized using DNA Polymerase I (New England BioLabs, M0209S). After enzyme inactivation and DNA purification with 1.5× volume of AMPure beads (Beckman Coulter, A63882), the DNA was subsequently fragmented by dsDNA Fragmentase (New England BioLabs, M0348S) at 37 °C for 15 min. The reaction was stopped by adding 5 μl 0.5 M EDTA and incubated at 65 °C for 15 min in the presence of 50 mM dithiothreitol. Next, the DNA was diluted to 40 μl with TE buffer and purified with 1.5× volume of AMPure beads. The eluted DNA was used for sequencing library preparation with NEBNext Ultra II DNA Library Prep kit (New England BioLabs, E7645). Biotinylated DNA fragments were enriched with 5 μl Dynabeads M-280 Streptavidin (Thermo Fisher, 11205D) and further amplified for 12 cycles by PCR.
Following PCR amplification, each amplicon was cleaned by 1× volume of AMPure XP beads twice and quantified with a Qubit 2.0 fluorometer (Invitrogen). The amplicon size and purity were further evaluated on an Agilent 2200 Tape Station (Agilent, 5067-5576). Equal amounts of amplicon were then multiplexed and sequenced using 2 × 150 cycles on an Illumina NextSeq 500 or NovaSeq 6000 platform (Rockefeller University Genomics Resource Center)36.
The SEnd-seq data were analysed and presented using custom analysis scripts from Ju and colleagues and is available in GitHub36.
LacZ and luciferase dual reporter
Mtb was cultured in standing 5 ml volumes in biological duplicate to an OD of 0.4–1.0. Fluorescein di-D-galactopyranoside (FDG) (Thermo Fisher, F1179), a cell permeable fluorescent LacZ substrate, was added to the cultures at a final concentration of 20 µM and incubated at 37 °C for 1 h. The Renilla Glo Assay kit from Promega (E2710) was used as instructed by the manufacturer. Briefly, after incubation, 5-ml cultures were spun down at ~3,900 × g for 10 min. Pellets were resuspended in 1 ml PBS and split into two samples for parallel LacZ and Renilla measurements. Samples were spun down at ~1,800 × g for 10 min, LacZ samples were resuspended in 155 µl PBS, and 50 µl was plated in triplicate in a 384-well plate. Luciferase samples were resuspended in 155 µl 1× assay buffer and plated in triplicate. Fluorescence was measured at an excitation of 485 nm and an emission of 515 nm, 30 flashes and an integration time of 40 µs in a TECAN plate reader. Luminescence was measured at an integration time of 5 ms using the TECAN luminescence setting. Background signal, if applicable, was subtracted from the values. For each sample, relative expression of luciferase (RLU) values were divided by relative expression of lacZ (RFU) values. This ratio was then divided by the same value of the no-terminator construct.
MIC 90 determinations
All compounds were dissolved in DMSO and aliquoted into a 384-well plate format using an HP D300e digital dispenser. Mtb cultures were grown to a late-logarithmic phase (OD 600 ≈ 0.8) and then back diluted to a starting OD 600 of 0.01. Of the diluted cell suspension, 50 µl was added in triplicate to wells containing the test and control compounds. Plates were incubated standing at 37 °C with 5% CO 2 . OD 600 was measured using a Tecan Spark plate reader at days 7, 10, 12 and 14 post plating, and percent growth was calculated relative to the DMSO vehicle control for each strain. MIC 90 measurements were calculated using a nonlinear fit in GraphPad Prism. For all MIC curves, data represent the mean ± s.e.m. for triplicate wells.
mScarlet reporter assays
mScarlet fluorescence was measured using a Tecan Spark plate reader at an excitation of 563 nm and emission of 600 nm. Mtb cultures were grown to a late-logarithmic phase (OD 600 ≈ 0.8) and then back diluted to a starting OD 600 of 0.01. A volume of 50 µl of the diluted cell suspension was added in triplicate to wells containing the test and control compounds. Plates were incubated under standing conditions and evaluated using the plate reader for fluorescence and optical density at days 5, 7, 12 and 14 post plating. Normalized fluorescence was calculated by dividing the background-adjusted fluorescence value by the background-adjusted optical density value.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.






© All Rights Reserved.