In cultured bacteria, this proportion varies widely, ranging from 20% up to 86% (Lobb et al.
Escherichia coli and Bacillus subtilis), as well as genome size: the larger the genome, the higher the proportion of genes with unknown functions.
More recent estimates have reduced this number down to 738, which still represent 15.5% of total genes (Moore et al.
Yet, they fail when no orthologs are present and when novel unknown functions are encoded.
The integration of multi-omics datasets offers another approach for the formulation of hypotheses on unknown gene function, linking changes across omics layers to single gene changes (e.g.
Throughout this Perspective, we use the term “unknome” to refer to protein-coding genes that simultaneously lack direct experimental evidence of a molecular or cellular role and lack informative homology to an already characterised protein. By this graded operational criterion (Vanni et al. 2022), a predicted domain of unknown function, an automatic or hypothetical annotation, or a context-dependent functional hypothesis may prioritise a gene but does not, by itself, remove it from the unknome: a gene leaves the unknome only when its function is supported by direct experimental validation or by confident homology-based transfer from a characterised protein. Genes which fulfil these criteria, and are then of unknown function, continue to represent a substantial fraction, between 40 and 60%, of the protein-coding genes identified in genomes and metagenomes (Vanni et al. 2022). In cultured bacteria, this proportion varies widely, ranging from 20% up to 86% (Lobb et al. 2020). This variability is largely a consequence of taxonomic bias where research efforts have been heavily concentrated on a few model organisms (e.g. Escherichia coli and Bacillus subtilis), as well as genome size: the larger the genome, the higher the proportion of genes with unknown functions. Notably, even in E. coli K12, one of the most studied bacterial models, ~1600 (~34% of total) genes lack experimental function evidence (“y-ome”), with 111 genes showing no functional clues at all (Ghatak et al. 2019). More recent estimates have reduced this number down to 738, which still represent 15.5% of total genes (Moore et al. 2024). For B. subtilis a similar fraction (~25% of total genes) is still unknown (Elfmann et al. 2025). Of course, we cannot exclude that factors such as functions not captured or propagated (“false unknown”) or non-homologous proteins performing the same function (non-orthologous displacements) may contribute to such a large fraction of still unknown function genes. Indeed, “false unknowns” are genes whose function is in fact known but was not captured or propagated during the annotation of a given genome. On the other hand, non-orthologous gene displacement occurs when a known function is carried out by a protein with no homology to the canonical one: such a protein lacks recognisable orthologs and is therefore flagged as “unknown”, even though the biochemical function it performs is already characterised.
Most genomic and metagenomic studies exclude this functionally uncharacterised fraction, limiting their findings to known, conserved pathways and essential housekeeping functions (Quince et al. 2017). This narrow focus presents a major bottleneck in genetics, microbiological and molecular biology research, as it risks creating an incomplete view of an organism’s biology based solely on existing knowledge.
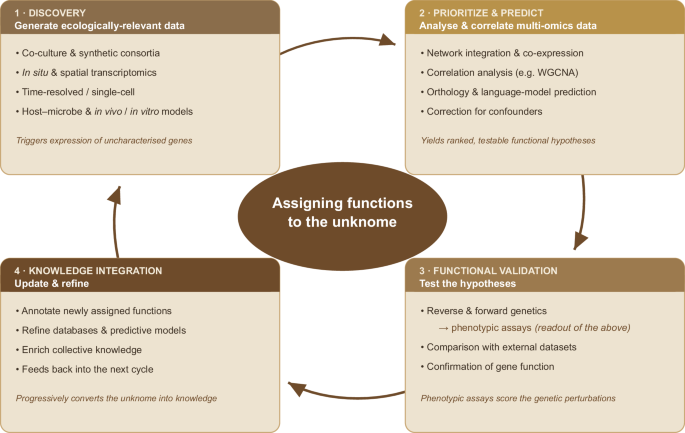
To address this gap, various experimental approaches and computational biology techniques have been proposed to functionally characterise these unknown genes, collectively referred to as the “unknome” (Rocha et al. 2023). In eukaryotic models, reverse genetics approaches such as RNA interference (RNAi) and CRISPR/Cas9 gene disruption have been instrumental (Rappsilber 2024; Rocha et al. 2023). In bacteria, forward genetics methods are more commonly used, including high-throughput techniques like transposon sequencing (Tn-Seq) (van Opijnen and Camilli 2013), which links gene function to an organism’s fitness under specific experimental conditions. Studies using these methods have successfully identified previously unknown function genes relevant to biological processes, critical for infectivity in pathogens such as Yersinia pestis and Dickeya dadantii (Eichelberger et al. 2020; Royet et al. 2019). At a much larger scale, a landmark survey using randomly barcoded transposon sequencing (RB-TnSeq) assigned mutant phenotypes, and thereby candidate functions, to thousands of genes of unknown function across dozens of diverse bacteria assayed under hundreds of conditions (Price et al. 2018).
Data-driven approaches rely on phylogenetic and computational methods to define orthology (Ribeiro et al. 2025). Yet, they fail when no orthologs are present and when novel unknown functions are encoded. Machine learning methods have also been employed, though a recent effort of reclassification of Enzyme Commission (EC) number predictions has shown a significant limitation of this method (Dias et al. 2025). More recently, machine-learning and language-model approaches have advanced rapidly: contrastive-learning models improve enzyme-function (EC) assignment (e.g. CLEAN; Yu et al. 2023), while mixed-modality genomic language models such as gLM2 exploit conserved genomic-neighbourhood context to retrieve functionally related genes that sequence-based search alone would miss (Jha et al. 2025). These methods substantially enlarge the space of testable functional hypotheses, yet remain least reliable for the genuinely novel, lineage-specific genes that dominate the unknome. The integration of multi-omics datasets offers another approach for the formulation of hypotheses on unknown gene function, linking changes across omics layers to single gene changes (e.g. fold change in gene expression) (Slobodyanyuk et al. 2024). For the well-studied model organisms, a recently developed resource, the Unknome v3 database (Rocha et al. 2023), systematically catalogues genes of unknown function, transforming neglected data into a potential source for new discoveries. However, equivalent resources are still largely missing for less-studied species.
Nevertheless, both these experimental and computational approaches have a common limit: they fail to capture the ecological relevance. Such experimental setups often rely on in vitro or simplified environments, such as pure cultures in synthetic media, which do not fully represent an organism’s ecological context. Similarly, computational methods based on sequence homology or automated annotation transfer often fail to capture functions that are lineage-specific or environment-dependent.
In contrast, recent approaches that incorporate ecological or contextual data are beginning to yield crucial insights into this hidden functional diversity. Another framework to explore the relevance of the unknome across organisms and environments has recently been developed, analysing a dataset of 71 million unknown genes from approximately 1700 metagenomes and 29,000 genomes (Vanni et al. 2022). This study found that the unknome is primarily lineage-specific and phylogenetically conserved, underscoring the importance of studying these unknown genes for insights into microbial diversification and niche adaptation, as also reported in other investigations (Coelho et al. 2022; Pavlopoulos et al. 2023). These recent studies pointed out that genomic context analysis and structural alignments enable predictions of functional associations for novel gene families found in environmental genomes (Rodríguez del Río et al. 2024). Many of these families are linked to biotic interactions, including antimicrobial resistance, antimicrobial peptide production, and secondary metabolite synthesis. This convergence of evidence suggests that the unknome is not merely ‘accessory’, but a rich reservoir of functions critical for ecological adaptation, particularly for the complex dialogues between organisms (Rocha et al. 2023).