
Mobile genetic elements shape microbial diversity and functions in thawing permafrost soils
MGE recombinase identificationRecombinase identification was on the basis of a framework recently developed to robustly identify and classify MGE recombinases from microbial isolate genomes15. Identified recombinases were assigned to the respective MGE recombinase type corresponding to the HMM with the best score, following previous recommendations15. Overall, 1.6% of the 62,660 CE-type recombinase genes identified by our analyses were considered near-complete CE by conjscan. Genomic neighbourhoods of MGE recombinases and mobility evaluationMGE mobility was assessed by comparing the genomic neighbourhoods of individual recombinases. MGE recombinase activity by metatranscriptomeMGE activity was assessed via documenting which MGE recombinases were detectably transcribed in metatranscriptomes.
Data collection, metagenome assembly and binning, gene annotation and taxonomy assignment
Our study site, Stordalen Mire, is a long-term ecological research site in northern Sweden. Large sampling and sequencing efforts since 2010 have yielded ~4.9 Tb of metagenomic sequence data from 615 readsets from 408 bulk soil samples that span space (across the site’s three habitat types: n = 188 palsa, n = 200 bog and n = 227 fen; average of ~8 Gb per sample) and time (summer peak growing season, 2010–2017) (Supplementary Table 1) with details described in another parallel study29, in previous studies26,27,50 and summarized below. The IsoGenie Consortium and EMERGE Biology Integration Institute have developed extensive resources from Stordalen Mire metagenome data, including assembly across all samples to produce 30,710,180 contigs (≥1,500 bp) and 36,419 medium- to high-quality MAGs (completeness ≥70% and contamination ≤10%; CheckM2 v1.0.2)51 that clustered into 2,048 populations (95% average nucleotide identity (ANI)) per community standards52. Select samples have also been sequenced using metatranscriptomics to assess gene expression (50 spatio-temporally diverse samples; average of ~18 Gb per sample). For this work, we additionally performed paired long- and short-read DNA sequencing on six samples, chosen to be representative of the larger dataset, as a control to assess short-read MGE recovery.
The protocol used for sample preparation and sequencing for short-read metagenomes has been described previously26,27,50. In brief, nucleic acids were extracted according to a published protocol53, except for the MAG source samples from substrate addition incubations (‘201607_SubstrateInc_9to19’), which were extracted with the DNeasy PowerSoil Kit (cat. no. 12988-100, Qiagen). Libraries were prepared with either KAPA Hyperprep, TruSeq or Nextera XT kits for short-read metagenomes; SMRTbell Express Template Prep Kit 2.0 for long-read metagenomes; or TruSeq Stranded mRNA kit for metatranscriptomes. Reads were sequenced with either Illumina HiSeq (2000 or 2500), NovaSeq 6000 or NextSeq 500 for short-read metagenomes; PacBio Sequel IIe for long-read metagenomes; or NovaSeq 6000 for metatranscriptomes.
Short-read sequencing data from individual samples were trimmed by Trimmomatic (v.0.36)54, assembled by SPAdes with the ‘--meta’ option (v.3.12)55 and then binned by MetaBAT256, GroopM257 and MaxBin258 (Cronin.v2), or an ensemble approach29. Those sequenced at Joint Genome Institute (JGI) were processed by JGI’s standard assembly and binning pipeline. In brief, reads were quality controlled, filtered and trimmed using the BBTools package59, assembled with SPAdes with the ‘--meta’ option55 and binned with MetaBAT256. The bins produced from this effort were combined with 9,406 bins from ref. 26 and 11,120 bins from stable isotope probing experiments with field peat soil and labelled litter (sequenced with NovaSeq at JGI and assembled with MEGAHIT v1.1.3)60 and then filtered using 70% completeness and 10% contamination CheckM2 (v1.0.2)51 cut-offs to form the 36,419 MAGs (consisting of 4,193 from Cronin.v1, 27,423 from Cronin.v2, 847 from JGI, 1,543 from ref. 26 and 2,413 from stable isotope probing). Next, bins obtained from the same metagenome were de-duplicated using Galah (95% ANI, commit e9e59a8)52, and all bins across the entire dataset were further dereplicated at 95% ANI using Galah (commit e9e59a8), producing 2,048 population level clusters. Further, the contigs encoding a recombinase were annotated with DRAM (–min_contig_size 1500)61 and taxonomically assigned at phylum level with CAT62, requiring a minimal contig length of 3 kb. The contigs in MAGs were assigned to the MAG taxon (obtained with GTDB-tk)63, as taxonomic assignments based on whole MAGs are typically more reliable than single-contig assignments.
MGE recombinase identification
Recombinase identification was on the basis of a framework recently developed to robustly identify and classify MGE recombinases from microbial isolate genomes15. Here, we applied the same recombinase HMM profiles and suggested cut-offs to identify MGEs in metagenomic assemblies. In brief, open reading frames (ORFs) were predicted from assembled contigs by prodigal (v2.6.3) using the ‘-p meta’ option64; then, recombinases were identified among the predicted protein sequences by hmmsearch (v3.3.2)65 using the 68 recombinase subfamily HMMs (Supplementary Table 2) and the ‘–cut-ga’ option. Identified recombinases were assigned to the respective MGE recombinase type corresponding to the HMM with the best score, following previous recommendations15.
Annotation-based inclusion/exclusion criteria
MGE recombinase candidates were further quality controlled, as recommended previously15, by using eggnog-mapper (v2.1.9, default settings)66. First, genes annotated as invertase, replication protein, DNA repair, DNA binding, homologous recombination, viral exonucleases or reverse transcriptase (interpreted to be spurious hits to other host genes related to DNA manipulation) were removed, except for the ones also annotated with the Pfam catalytic domains of ‘transposase’ or ‘helicase’ that were retained after manual curation. This removed 0.68% of the identified possible recombinases (Supplementary Fig. 1). Second, sequences annotated by eggnog-mapper as CRISPR, defense response to virus or maintenance of DNA repeat elements were removed to filter out spurious hits thought to be similar to Cas family recombinases, but not considered as an MGE recombinase. This removed 0.46% of the identified possible recombinases. Third, we discarded predicted recombinases that contained only a helix–turn–helix (DNA binding) domain, as well as relaxase hits for which Pfam annotates a helix domain, but without the catalytic domain required for recombinase activity. This removed 0.07% of the identified possible recombinases. Fourth, recombinases matching a ‘cell’ type recombinase HMM (and thus unlikely to be encoded by an MGE) were removed. This removed 2.52% of the identified candidate recombinases, and yielded a final total of 2,107,204 MGE recombinases (Supplementary Table 3, item A). Last, some recombinase subfamilies/HMMs include recombinases from different MGE types, which prevents assignment for that HMM to a specific MGE type. These recombinases with ambiguous origins accounted for 20.51% of the identified recombinases. They were included in the analyses of total MGE recombinases, but excluded in analyses of individual MGE types, which focused on four recombinase-based MGE types: ‘IS_Tn’ (insertion sequence and transposon), ‘CE’ (conjugative elements including conjugative element and mobility islands), ‘phage’ (phage and phage-like elements) and ‘integron’ (integron) (Supplementary Fig. 1). Note that IS_Tn recombinases integrated into the host chromosome, as opposed to into larger MGEs, cannot be distinguished using our approach and data, as defining the boundaries of the MGE is challenging in short-read assemblies, except for a small fraction of MGEs with curated boundaries (see below). To assess the diversity of these MGE recombinases, we considered recombinase sequences as ‘redundant’ when they clustered with other sequences at 100% AAI using CD-HIT (v4.8.1)67 with ‘-c 1 -g 0 -G 1 -l 30 -d 0 -M 0’. This yielded 1,130,776 clusters that we considered as unique recombinases.
Identification of cargo genes/interrupted genes associated with MGEs
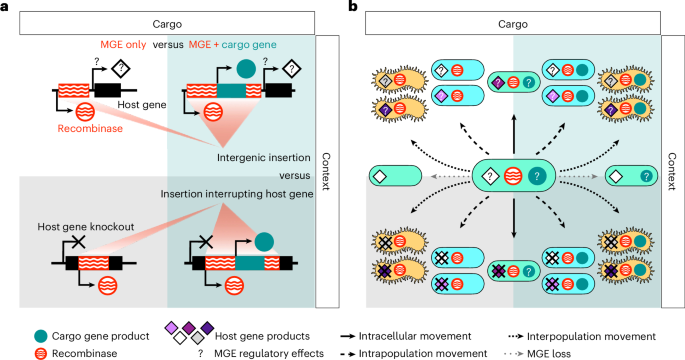
Once MGE recombinases were identified, we surveyed their genomic context to assess potential cargo and/or interrupted genes. This was done using MGE-specific approaches as follows.
Integron
All integron-containing contigs were screened for cargo genes (also known as ‘gene cassettes’) using integron-finder (v2.0.2)68 with default parameters. In brief, integron-finder identifies integrons by using integron-specific integrase HMMs to identify integron integrases (intI), and a covariance model to identify the integron attachment site (attC), as the two bounding regions of an integron sequence. Only integrons encoding intI and at least one attC were treated as ‘complete’ by integron-finder and were retained. There are two types of integron structures that are known in terms of the orientation of intI and attC: (1) a regular type with intI and attC on opposite strands and facing outwards and (2) another type with intI and attC on the same strand, called an inverted integrase integron68. Of the 686 ‘complete’ integrons we identified, 681 integrons also met these orientation expectations and the 5 that did not were not considered further. The gene cassettes delimited by intI and attCs in these integrons were treated as cargo genes associated with integrons69, resulting in integron regions with various lengths (minimum 983 bp, median 2,310 bp, mean 3,092 bp and maximum 14,195 bp). Problematically, most contigs (97.4% or 25,953 out of 26,634) encoding Khedkar’s ‘integron’ type recombinases were not considered complete by integron-finder. To test this, we also replaced the integron integrase HMM in the integron-finder tool with that from the study by Khedkar et al.15 and found it produced nearly identical results and did not yield additional instances of predicted complete integrons. One explanation could be that attC, not integron integrase, is the limitation, as the flanking attC sequences are identified using a database of known attC sequences and these could miss novel attC sequences used by integrons in natural microbiomes. Given this, we interpret the large discrepancy between Khedkar’s and integron-finder’s integrons to represent one of the following scenarios: (1) degraded or domesticated integrons that lack attC sites70,71, (2) integrons that lack attC sites owing to fragmented assembly or (3) intact and probably complete integrons that have novel attC sites. Thus our analyses will underestimate the extent of cargo genes carried by integrons in Stordalen Mire.
CE
CEs are made of three key components—a relaxosome (includes the CE-type recombinase), a conjugation couple protein and a type IV secretion system (T4SS)—that can be used as hallmark genes that are more conserved than other CE genes72. To identify cargo genes associated with CE, the CE hallmark genes were identified in CE recombinase-encoding contigs by conjscan implemented in MacSyFinder (v2.0rc7) with ‘–models CONJScan/Plasmids all–db-type gembase’ and ‘–models CONJScan/Chromosome all–db-type gembase’ with gene protein sequences as input72,73. Conjscan specifically establishes eight types of CEs using hallmark genes, which are core across all eight CE types, and accessory genes that are diagnostic for specific CE types. CEs identified by conjscan from Stordalen Mire sequence data were those that matched a relaxase, a couple protein (an ATPase that couples MGE with the T4SS), a T4SS ATPase and an additional structural gene from T4SS. For the purposes of identifying cargo genes, matches of one model, MOB, were removed, as it only requires the relaxase and thus does not identify the boundaries to establish a complete CE. As a result, we identified 1,017 near-complete CEs. As recommended, we required the distance between the hallmark and accessory genes to be ≤30 genes, except for the relaxase, which could be as much as 60 genes away from any other hallmark and accessory genes72. In a small number (13 of 1,017) of instances conjscan identified >1 CE in a region. For these, we manually separated the identified hallmark and accessory genes into subgroups according to gene proximity and required each subgroup to include all hallmark genes (relaxase, conjugation couple protein and T4SS ATPase) and at least one accessory T4SS gene. Once such ‘complete’ CEs were identified, the genes between the leftmost and rightmost hallmark and accessory genes were assumed to be cargo genes carried by the CE (except for hallmark and accessory genes themselves). This resulted in CE regions with various lengths (minimum 7,907 bp, median 16,290 bp, mean 23,383 bp and maximum 81,148 bp). Overall, 1.6% of the 62,660 CE-type recombinase genes identified by our analyses were considered near-complete CE by conjscan. Again, this was not a function of improved HMMs as we tested replacing the relaxase HMMs in conjscan by those from Khedkar et al.15 and found it produced similar results. Instead we interpret this large discrepancy to suggest that most CEs in these data are degraded, fragmented from poor assembly or cannot be formally detected as complete because of limitation and bias in hallmark gene databases.
Insertion sequences/transposons
To identify IS_Tn recombinase-interrupted genes, up to 3,000 bp upstream and downstream of IS_Tn recombinases were collected from each contig, requiring a minimal length of at least 50 bp on each end and at least 500 bp across the two ends combined (with 1,063,800 IS_Tn recombinases in total meeting the requirements), and then aligned by minimap2 (v.2.22, default settings)74 to the coding sequence of representatives of protein clusters (PCs) obtained by MMseqs2 at 95% identity (v4.8.1)75 from all contigs (≥1,500 bp) used in this study. The corresponding coding sequence was considered interrupted if the following four conditions were met: (1) if upstream and downstream alignments combined covered ≥50% of the coding sequence of the reference PC; (2) if there is a gap between upstream and downstream alignments of the coding sequence, the gap is ≤10% of the coding sequence length; (3) if there is an overlap between upstream and downstream alignments on the coding sequence, the overlap is ≤10% of the shorter alignment; and (4) if there are multiple alignments either upstream or downstream of the coding sequence, the pair with largest coverage of coding sequence is chosen. This identified 5,342 PCs putatively interrupted by a total of 12,332 IS_Tn-type recombinases.
Phage/phage-like elements
All contigs encoding ‘phage’ recombinases were screened for temperate phage by VirSorter2 (v2.2.3) following VS2 SOP step 1 (v3.0)76,77, and the predicted phage sequences trimmed by checkV (default setting, version 0.7.0)78. Next, the checkV trimmed sequences encoding a ‘phage’ recombinase were considered as phage/phage-like regions. To identify cargo genes, that is, host(-like) genes encoded on phage/phage-like elements, we opted for a conservative approach in which we reduced the dataset for ‘cargo gene inspection’ to include only phage/phage-like regions with one ‘phage’ recombinase, and use the leftmost and rightmost checkV, called ‘viral gene’ or ‘phage’ recombinases, as the boundaries. This identified 4,277 temperate phages with ‘phage’ recombinases and at least one extra gene other than the two at the ends. Meanwhile, 347 phage/phage-like elements were removed for having more than one ‘phage’ recombinase, in order to avoid cases where multiple contiguous temperate phages or degraded ones yielded a single prophage prediction, which could lead to host genes being mistaken as cargo genes. This resulted in 3,930 conserved phage/phage-like regions (minimum 1,386 bp, median 7,293 bp, mean 13,779 bp and maximum 352,302 bp).
Annotations of cargo/interrupted genes
Insertion sequence or transposon interrupted genes and genes within other MGEs were functionally annotated using KEGG and Pfam databases in DRAMv for phages/phage-like elements or DRAM for other MGEs (v1.2.3,–min_contig_size 1500)61, and further evaluated by comparison with best hits in NCBI NR (downloaded on 4 October 2020) using DIAMOND (v2.0.8.146, blastp bitscore ≥50)79. A subset of the genes of specific interest to the Stordalen Mire thawing permafrost system were selected and manually curated for display (Supplementary Table 4) including transport, carbon/nitrogen/sulfur cycling, signalling, antiviral defense, salt stress response and genome maintenance and expression.
Evaluation of short-read assembly and binning artefacts for recovering MGE recombinases using long reads
Assembly and binning of paired long reads and short reads
Soil samples (six total) were collected at the Stordalen Mire site from 2019 from two depths (centimetres below ground) across three habitats (palsa, bog and fen). DNA extraction and Illumina NovaSeq sequencing was performed as described above. In addition, DNA sequencing was performed from the same DNA extractions using the PacBio Sequel IIe HiFi sequencing platform at JGI. PacBio data were processed at JGI to form filtered circular consensus sequencing reads. Assemblies were generated through two methods: short-only and hybrid short-read plus long-read (herein, hybrid). Short-only was assembled with metaSPAdes v3.15.455 using Aviary v0.5.380 with default parameters. Hybrid assembly was performed using Aviary v0.5.3 with default parameters. This involved a step-down procedure with long-read assembly through metaFlye v2.9-b176881, followed by short-read polishing by Racon v1.4.382, Pilon v1.2483 and then Racon again. Next, reads that did not map to high-quality metaFlye contigs were hybrid assembled with metaSPAdes v3.15.4 and binned out with MetaBAT2 v2.1.556. For each bin, the reads within the bin were hybrid assembled using Unicycler v0.4.884. The high-coverage metaFlye contigs and Unicycler contigs were then combined to form the assembly fasta file. Genome recovery was performed using Aviary v0.5.3 with samples chosen for differential abundance binning by Bin Chicken v0.4.285 using SingleM metapackage S3.0.586. This involved initial read mapping through CoverM v0.6.187 using minimap2 v2.1874 and binning by MetaBAT v2.1.56, MetaBAT2 v2.1.556, VAMB v3.0.288, SemiBin v1.3.189, Rosella v0.4.290, CONCOCT v1.1.091 and MaxBin2 v2.2.758. Genomes were analysed using CheckM2 v1.0.251 and clustered at 95% ANI using Galah v0.4.052.
Assessment of MGE recombinase recovery by short-read assembly and binning
We considered the high-quality hybrid MAGs as the best available references and used these to assess the recovery of MGE recombinases in short-read contigs and MAGs. A total of 17 high-quality MAGs (completeness ≥95%, contaminant ≤5% and at most 100 contigs) from hybrid assembly were selected and paired with 17 MAGs recovered from short-read assembly through the 95% ANI clusters (the highest ANI were chosen if there were many) for comparison. The short-read MAGs included 5 medium quality (≥70% completeness, ≤10% contamination) and 12 high quality (≥95% completeness, ≤5% contamination).
Evaluation of these 17 MAGs were carried out as follows. First, each pair of hybrid MAG and short-read MAG were compared in terms of total number of MGE recombinases, as well as unique and shared MGE recombinases at 100% AAI by CD-HIT (v4.8.1) with ‘-c 1 -g 0 -G 1 -l 30 -d 0 -M 0’ (ref. 67). Second, we evaluated the impact of sequencing depth, assembly and binning to assess which step was most problematically leading to under-detection of MGE recombinase identification in short reads. Initially, we mapped short-read assembled contigs (sheared into 500-bp fragments) to hybrid MAGs from the same sample using CoverM v0.6.1, requiring each read to be 75% aligned with 95% identity. Third, these data were used to assign MGE recombinases in the 17 hybrid MAGs into one of three categories: (1) those recovered by short-read MAGs (termed ‘covered’); (2) those not fully covered by short-read contigs, that is, those missed by short-read assembly (termed ‘missed assembly’); and (3) those fully covered by short-read contigs, that is, those recovered by short-read assembly but missed by short-read binning (termed ‘missed binning’). Fourth, the coverage of each gene was generated by dirseq v0.4.326 using the BAM file from above and used to assess the relative abundances of each category as a metric for the impact on MGE-representation in the short-read assemblies.
With a better understanding for which step was failing most often for MGE recombinase recovery in short-read assemblies, we sought next to evaluate what factor(s) might be causing the failed capture events. First, we evaluated sequencing depth, as low coverage might cause breakages in short-read assembly graphs. This was done by mapping trimmed short reads to hybrid MAGs using CoverM v0.6.1, requiring each read to be 75% aligned with 95% identity, calculating the read coverage of each gene by dirseq v0.4.3 and comparing coverages among the three categories of MGE recombinases. Second, we compared the genomic neighbourhood number among the three categories, reasoning that perhaps indels were an issue. Genomic neighbourhood number was calculated by clustering genomic neighbourhoods of each unique MGE recombinase (see below) at 90% ANI. To test this, nucleotide diversity for each base position was calculated as 1 − [(number of ‘A’ bases/total bases)2 + (number of ‘C’ bases/total bases)2 + (number of ‘T’ bases/total bases)2 + (number of ‘G’ bases/total bases)2 + (number of ‘deletion’ bases/total bases)2 + (number of ‘insertion’ bases/total bases)2], adopting the nucleotide diversity definition from inStrain7 with the addition of deletion and insertion variants. The variant count at the base position was generated by pysamstats (v1.1.2; https://github.com/alimanfoo/pysamstats) with the ‘–type=variation’ option. Third, we assessed the nucleotide diversity of each MGE recombinase, reasoning that perhaps elevated nucleotide diversity could lead to short-read assembly graph breakages. To test this, for each of the 17 MAG pairs, MGE recombinases only identified in hybrid MAG were compared with short-read assembled contigs to track whether they were assembled by short reads, and then whether they were binned in MAGs from other MAG clusters. The tracking of MGE recombinases in contigs was accomplished with ‘MMseqs map’ (v13.45111), with default settings, and then filtered for hits with 100% identity in the protein sequence75.
MGE recombinase per genome estimation from contigs and MAGs
To estimate the MGE recombinase number per genome from contigs, the median count of ribosomal genes was first used to estimate the number of genomes in each assembly. To do this, ribosomal genes were extracted from contig annotations by DRAM61 using KEGG IDs (Supplementary Table 6), and the average number of recombinase genes per genome was calculated by dividing the total number of recombinases in the sample by the estimated total number of genomes. We next linked individual recombinase-encoding contigs to MAGs built from the same sample. To that end, we used exact string searching to identify MGE-recombinase-encoding contigs in the dereplicated MAGs from the same sample. Through this process, 106,017 out of a total 665,700 (15.9%) MGE-recombinases-encoding contigs ≥3 kbp could be linked to a MAG. To account for partial MAG recovery, the number of MGE recombinases per genome estimated from MAGs was calculated as the number of MGE recombinases in a given MAG divided by MAG completeness.
Genomic neighbourhoods of MGE recombinases and mobility evaluation
MGE mobility was assessed by comparing the genomic neighbourhoods of individual recombinases. For this analysis, we limited the input data to recombinases that occurred ≥2 times in the total dataset (assembled contigs per sample). This was achieved by clustering the recombinases at 100% AAI using CD-HIT v4.8.1 with ‘-c 1 -g 0 -G 1 -l 30 -d 0 -M 0’ (ref. 67), only keeping the clusters with at least two recombinases. For each MGE recombinase, 300 bp upstream and downstream of the recombinase were considered as its immediate genomic neighbourhoods, excluding those near the contig ends (<300 bp). For each recombinase cluster, a pairwise comparison of neighbourhoods was performed among all member recombinases using both neighbourhoods (that is, upstream versus upstream and downstream versus downstream) using BLAST+ (v2.8.1+) with ‘-task blastn’92 and a bitscore ≥50. The ANI for each neighbourhood comparison was calculated as the ‘sequence identity from blastn output’ multiplied by the ‘alignment coverages’. For each comparison, we labelled the neighbourhood with the higher ANI value as ‘high_ani’ and the neighbourhood with lower ANI as ‘low_ani’. For some MGEs, we expect the neighbourhood on the MGE side of the recombinase to be part of the MGE, and thus remain identical after integration into a new locus, so we used ‘low_ani’ values (that is, the lower ANI between the upstream and downstream region of the pair) to evaluate whether the neighbourhood of an MGE changed or not. An MGE recombinase pair was considered to be in different genomic locations if ‘low_ani’ was <1%, and a recombinase cluster was treated as active if one pairwise comparison showed different neighbourhoods. This approach only used immediate neighbourhoods (within 300 bp) as a requirement, as neighbourhoods with longer ranges would remove more recombinases from this analysis owing to the short average contig length. However, as a sensitivity analysis, we also tested the above with 600-bp and 1,000-bp neighbourhoods. These analyses showed that the size of the genomic neighbourhood (300–1,000 bp) had very little impact on the fraction of MGE recombinases identified as mobile via this changing genomic neighbourhood metric (Extended Data Fig. 9).
MGE recombinase activity by metatranscriptome
MGE activity was assessed via documenting which MGE recombinases were detectably transcribed in metatranscriptomes. Leveraging 81 paired metatranscriptomes and metagenomes, we evaluated MGE recombinase transcriptional activity by mapping metatranscriptome reads to contigs assembled from metagenomes using transcriptM v0.3.193 with ORF predictions from DRAM (see above) as the ‘–gff’ input, which also removed tRNA, rRNA, tmRNA and PhiX from the reads. To ensure the quality of RNA sequencing data (reverse stranded), we only analysed 58 pairs with ≥80% of reads mapped to the coding reverse strand of ORFs in contigs, based on dirseq v0.4.326 with BAM and GFF files from above as inputs (Supplementary Table 5). We only considered contigs ≥10 kbp to avoid spurious ORF predictions from short contigs. Next, we defined genes with ≥90% of gene length covered as expressed using pydirseq v0.1.0-alpha with BAM and GFF files from above as inputs94. To normalize for the differences in sequencing depth and overall activity of the microbial community across samples, we used the average percentage of ribosomal genes (Supplementary Table 6) detected as active as the baseline activity to normalize other genes (termed ‘normalized active ratio’), as ribosomal genes are expected to be constitutively expressed in active cells. This ‘normalized active ratio’ thus provides an estimation of the number of genes detected as expressed within a functional category (for example, recombinases) relative to an estimated total number of genomes detected as transcriptionally active in the same sample.
MGE recombinase partial carriage by host population
As a result of MGE movement within the host population and an equilibrium of the co-evolution of MGEs and their hosts95, we noticed that some MGE recombinases were present in only a fraction of the contigs assigned to a given host population (termed ‘partial carriage’). To quantitatively evaluate this, trimmed metagenomic reads were mapped to contigs from the same sample using CoverM v0.6.1 with default settings87, the resulting BAM file was then filtered (requiring ≥90% of each read aligned with ≥95% identity) and used as the input for ‘samtools depth’ (v1.17) with the ‘-a’ option to get the read depth coverage at each position96. The first and last 150 bp of each contig were not considered, as read depth coverage near the ends are expected to be artificially lower. We defined partial carriage as cases where the coverage of the MGE recombinase was at least one standard deviation lower than the larger one (requiring a minimal coverage of 1×) of the average coverages of the upstream and downstream contig regions (Cohen’s d ≥ 1). To ensure enough host regions were considered for reliable mapping-based estimation of genome coverage, the MGE recombinase total set was filtered to 23,647 MGE recombinases with ≥20 kb upstream and downstream. Note that sensitivity analyses revealed similar patterns when analysing 2.5-kbp instead of 20-kbp regions (Extended Data Fig. 10), which suggests that this conservative cut-off did not substantially influence the detection of partial carriage in these data.
We expect that this method should give us a lower-bound estimate of partial carriage, as it is not able to detect cases for which (1) the hosts have low read coverage (rare), (2) the carriage ratio is lower than but close to 1, (3) host genome coverage has a wide range of variation in different genomic locations due to strain variation within the population or (4) the MGE recombinase is on the edge of a contig.
Statistics
All statistics and visualization were performed in R v4.2.197. The R packages and versions used are available via GitHub98. For statistical significance between groups, the Wilcoxon rank-sum test was used for two groups and Kruskal Wallis test was used for >2 groups as implemented in ‘compare_means’ in ggpubr package v0.6.0, with P values adjusted by Benjamini–Hochberg adjustment for multiple comparisons.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.







© All Rights Reserved.