
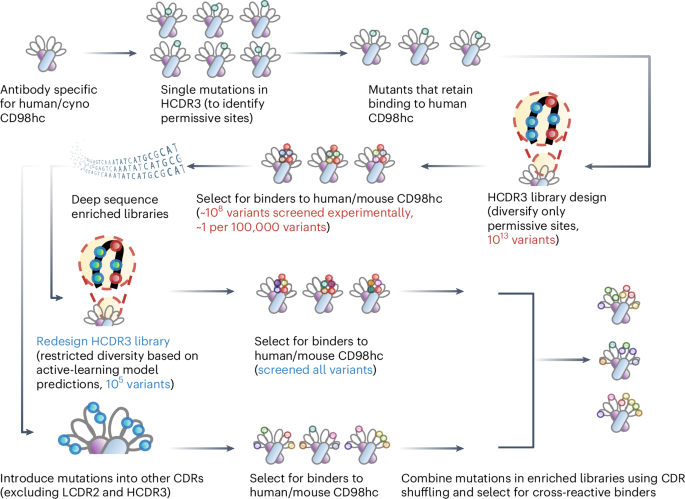
CD98hc-targeted antibody shuttles for central nervous system delivery with broad cross-species reactivity
Next-generation sequencing (NGS) analysis was performed on the sequences from HCDR3-I (MACS #3 output of HCDR3 library against mouse CD98hc), HCDR3-D1/D2 (FACS #1/#2 output against both mouse and human CD98hc), HCDR3-M1/M2 (FACS #1/#2 output against mouse CD98hc) and HCDR3-H1/H2 (FACS #1/#2 output against human CD98hc). For samples involving FLAG-tagged human CD98hc ectodomains, anti-rabbit Fc antibody conjugated to Alexa Fluor 568 (Invitrogen, A11011; 1:200) was also included. Following incubation, cells were washed again, pelleted and sorted for binding to mouse and/or human CD98hc using a Sony MA900 cell sorter. Binding signal of each mutant to CD98hc was normalized by WT antibody binding to human CD98hc. In this context, the epitope is defined as amino acids in human CD98hc within 4 Å of any binder atom, and the paratope as amino acids in the binder within 4 Å of any atom in human CD98hc.
Ethics statement
Animal studies were conducted following guidance for the care and use of laboratory animals as adopted by the NIH, under protocols PRO00010991, PRO00009238 and PRO00010447 approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Michigan.
Cloning of antibody and antigen genes
For yeast display, the variable heavy (V H ) and light (V L ) domains of CP1 (refs. 27,68) (GenBank ID OR253074.1), WT31 (PDB ID 7DF1) and selected variants were either isolated from yeast plasmids or synthesized as gene fragments (IDT). The V H and V L domains were cloned into pD208 yeast display vectors in the scFv format, which was Aga2-linker-V H -linker-V L for WT and variants thereof, and Aga2-linker-V L -linker-V H for CP1.
For producing soluble bispecific antibodies, three pTT5 mammalian expression vectors were used that encoded for a phosphorylated tau-targeting IgG with a CD98hc-targeting scFv fused to one of the C termini of the heavy chains (GenBank ID OR253074.1). These vectors corresponded to the light chain, heavy chain #1 (hole chain) and heavy chain #2 (knob chain) of human IgG1 antibodies. Heavy chain #2 contained knob mutations [S(365)C and T(377)W]48 and an scFv attached to the C terminus. Heavy chain #1 contained hole mutations [Y(360)C, T(377)S, L(379)A and Y(418)V]48. The variable regions of various scFvs, including CP1, WT and evolved variants thereof, were PCR amplified from yeast plasmids and cloned into heavy chain #2.
For producing CD98hc antigens, gene fragments were synthesized (IDT) and cloned into pTT5 mammalian expression vectors with an N-terminal 6×His tag.
DNA fragments were inserted into restriction enzyme-digested vectors as described in previous work27. Constructs were transformed into E. coli DH5α and verified by Sanger sequencing.
Recombinant antigen production
The HEK293-6E cells (National Research Council Canada, L-11565) was maintained in F17 medium (Thermo Fisher, 50591354) at 37 °C with 5% CO 2 and 250 r.p.m. shaking until reaching a density of 2 × 106 cells per ml. For transfection, 15 µg plasmid of recombinant CD98hc ectodomain was mixed with 45 µl 40 kDa polyethylenimine (1 mg ml−1, PEI MAX; Polysciences, 247651) in 3 ml culture medium, incubated for 15 min at room temperature and added to 25 ml cultures of HEK293-6E cells. At 24 h post transfection, 0.75 ml 20% (w/w) yeastolate (Thermo Fisher, B92804) was added. Cells were collected 6 d after transfection at 3,500 g for 40 min. The supernatant was filtered (Thermo Fisher, 166-0045) for downstream purification.
Ni-NTA agarose beads (Qiagen, 30250) were incubated with supernatants containing recombinant CD98hc overnight at 4 °C. Collected beads were washed three times with 10 ml PBS and incubated with 50 mM imidazole (pH 7.4) for 15 min as a wash step. The beads were then eluted by 500 mM imidazole (pH 7.4) for 15 min, with centrifugation at 1,000 g for 2 min after each step. The elute was buffer exchanged into PBS (pH 7.4) using desalting columns (Thermo Fisher, 89891) and stored at −80 °C. Protein concentration was determined by NanoDrop absorbance at 280 nm, and purity was evaluated by size-exclusion chromatography using a Superdex 200 Increase 10/300 GL column (Cytiva, 28990944). CD98hc preparations with purity <90% monomer underwent additional size-exclusion chromatography-based purification to ensure high purity. All samples after 2-step purification were assessed by SDS–PAGE.
Biotinylation of recombinant CD98hc ectodomains
Filtered PBS (180 μl) was added to 1 mg pre-aliquoted biotin (Fisher Scientific, A39257) to create a 10 mM stock. Proteins were incubated with biotin at a 20:1 molar ratio (biotin:protein) for 1 h at room temperature. Excess biotin was then quenched with 1.5 M hydroxylamine (10:1 molar ratio to biotin) for 1 h at room temperature. Proteins were aliquoted and stored at −80 °C until needed.
Histidine scanning analysis
Histidine scanning mutagenesis was performed on 16 residues (Kabat numbering) in HCDR3 using primers designed to replace the wild-type codons with histidine codons. Each mutant was cloned following the protocol in the cloning section, and sequence-verified plasmids were transformed into Saccharomyces cerevisiae EBY100 cells as described below.
Yeast cells were cultured overnight in 5 ml YPD medium (10 g l−1 yeast extract, 20 g l−1 bacto peptone, 20 g l−1 dextrose) at 30 °C. For each variant, 500 µl of cells were pelleted at 16,000 g for 1 min and resuspended in 500 µl ice-cold plate solution buffer (400 g l−1 PEG (molecular weight, 3,350), 6.6 g l−1 LiAc, 0.4 mM EDTA and 10 mM Tris HCl, pH 7.5) with 10 µl 100 ng µl−1 plasmid DNA and 10 µl 10 mg ml−1 single-stranded DNA (Sigma-Aldrich, D7656). The mixture was incubated on ice for 15 min, followed by a 15-min heat shock at 40 °C. Cells were pelleted, resuspended in 200 µl of deionized water, plated on dropout plates (15 g l−1 agar, 20 g l−1 dextrose, 6.7 g l−1 yeast nitrogen base (without amino acids), 3.8 g l−1 tryptophan dropout media) and incubated at 30 °C for 2 d. Single colonies were then cultured in 5 ml yeast minimal media (SDCAA (16.75 g l−1 sodium citrate, 4 g l−1 citric acid (anhydrous), 6.7 g l−1 yeast nitrogen base (without amino acids), 5 g l−1 acid casein peptone (casamino acids), 20 g l−1 dextrose)) at 30 °C and 225 r.p.m. for 2 d. Cells were then transferred from SDCAA to a yeast induction medium (SDGCAA; 6.76 g l−1 sodium phosphate dibasic dihydrate, 8.56 g l−1 sodium phosphate monobasic monohydrate, 6.7 g l−1 yeast nitrogen base (without amino acids), 5 g l−1 acid casein peptone (casamino acids), 20 g l−1 galactose, 2 g l−1 dextrose) at a starting optical density at 600 nm (OD 600 ) of 0.5–1.0 and grown for 48 h at 20 °C or 24 h at 30 °C. All media contained 100 µg ml−1 ampicillin, 100 µg ml−1 kanamycin, 100 µg ml−1 streptomycin and 100 units ml−1 penicillin.
Yeast cells were collected and washed twice with phosphate-buffered saline with 0.1% BSA (PBSB; 100 ml 10× PBS, 900 ml deionized water and 1 g BSA), aliquoted into a 96-well plate at a density of 1 × 105 cells per well. The cells were incubated in PBSB containing: 1:1,000 dilution of mouse anti-myc tag antibody (Cell Signaling Technology, 2276S), 1% milk and 100 nM of biotinylated mouse or human CD98hc protein at room temperature for 3 h. Then the cells in each well were washed with cold PBSB, resuspended and incubated on ice for 4 min in 200 µl of PBSB containing 1:1,000 dilution of streptavidin Alexa Fluor 647 (Invitrogen, S32357) and 1:200 dilution of anti-mouse Fc antibody conjugated with Alexa Fluor 488 (Invitrogen, A11001). Following incubation, cells were washed and resuspended in 150 µl cold PBSB per well. Samples were analysed using a Bio-Rad ZE5 cell analyser, and the resulting data were processed and visualized using GraphPad Prism.
Yeast library construction
For the initial HCDR3 library, 10 sites (H96, H97, H98, H99, H100, H100B, H100C, H100D, H100E, H102) in HCDR3 of the WT antibody were selected and subjected to soft mutagenesis42. This approach sampled the WT residue with a ~50% probability and the remaining 19 amino acids with a ~50% probability (theoretical diversity of ~1.0 × 1013).
For the redesigned HCDR3 library, the same 10 HCDR3 sites were targeted for mutagenesis on the basis of PSERM metrics to introduce specific substitutions. Next-generation sequencing (NGS) analysis was performed on the sequences from HCDR3-I (MACS #3 output of HCDR3 library against mouse CD98hc), HCDR3-D1/D2 (FACS #1/#2 output against both mouse and human CD98hc), HCDR3-M1/M2 (FACS #1/#2 output against mouse CD98hc) and HCDR3-H1/H2 (FACS #1/#2 output against human CD98hc). The average PSERM score for the 10 HCDR3 sites was calculated across all sequences on the basis of the PSERM metrics derived from HCDR3-D1 and HCDR3-I. In addition, the average PSERM score was determined for the 53 individual clones, and variants with PSERM scores >0.169 were considered to be species cross-reactive, defined as clones that have binding >4% to mouse and human CD98hc.
Sequences from all libraries with an average PSERM score > 0.169 were further analysed to evaluate amino acid distributions at each mutation site (Supplementary Table 2). On the basis of this analysis, mutagenic primers (Supplementary Table 3) were designed to construct a redesigned HCDR3 library. Degenerate codons were selected to preferentially encode amino acids corresponding to a cumulative probability of ~90% per position, with limited redundancy incorporated at specific sites to minimize primers number (Supplementary Table 4, theoretical library diversity of ~3.0 × 105). The reconstructed HCDR3 library was subsequently characterized by NGS (Supplementary Table 5).
Two affinity maturation libraries were generated for V H (HCDR1 and HCDR2) and V L (LCDR1 and LCDR3). Residue selection was guided by the crystal structure of the human CD98hc-WT antibody complex (PDB 7DF1). Distances were calculated between the Cα atoms of residues in the WT antibody CDRs and the corresponding epitope residues on human CD98hc (loop 1 and 2). LCDR2 was excluded due to its greater distance from the epitope. Residues from the four CDR loops with a minimum distance to the epitope of <5 Å were selected. Other residues with a minimum distance to the epitope between 5–8 Å were also selected if their side chains pointed towards the epitope, as determined by a Cβ (antibody)–epitope distance at least 0.3 Å shorter than the corresponding Cα (antibody)–epitope distance (highlighted in red in Supplementary Tables 6 and 7).
For the heavy chain library, antibody D3, derived from the initial HCDR3 library, served as the starting point. Eight HCDR1 and HCDR2 residues (H33, H35, H50, H52, H53, H54, H56, H58) were selected for soft mutagenesis (among them, residue H35 was incorporated as it is located at the interface of the HCDR1, HCDR2 and HCDR3, as shown in Supplementary Fig. 7), yielding a theoretical diversity of ~2.6 × 1010. For the light-chain library, D3 also served as the starting point. Nine LCDR1 and LCDR3 residues (L27D, L27E, L27F, L28, L32, L91, L92, L93, L94) were targeted by soft mutagenesis, yielding a theoretical diversity of ~5.1 × 1011. Finally, the three enriched libraries (HCDR1/HCDR2, LCDR1/LCDR3 and redesigned HCDR3) were combined using CDR-swapping mutagenesis44,45.
To transform each library, EBY100 yeast cells were grown in 5 ml YPD media overnight at 30 °C and 225 r.p.m., expanded into 50 ml YPD overnight under the same conditions and inoculated to 100 ml YPD at a starting OD of ~0.3. Cultures were grown at 30 °C and 225 r.p.m. until the OD 600 reached ~1.6. For each library, 50 ml EBY100 was pelleted at 2,500 g for 5 min at 4 °C, washed sequentially with 25 ml ice-cold sterile deionized water and 25 ml ice-cold electroporation buffer (1 M sorbitol, 1 mM calcium chloride), centrifuging after each step. Cells were then resuspended in 25 ml conditioning buffer (0.1 M lithium acetate, 10 mM 1,4-dithiothreitol), incubated for 15 min at 30 °C and 225 r.p.m., pelleted, washed once with 25 ml of ice-cold electroporation buffer and then resuspended in electroporation buffer to a final volume of ~400 μl. Ethanol-precipitated DNA was added to the cells and mixed until homogeneous. Cells were transferred to pre-chilled 2-mm gap electroporation cuvettes (Fisher Scientific, FB102) and electroporated using a Bio-Rad electroporator (Bio-Rad Gene Pulser Xcell) at 2,500 V, 200 Ω and 25 μF. Libraries were immediately recovered in 8 ml recovery media (1:1 mixture of YPD and 1 M sorbitol mixture) for 1 h at 30 °C and 225 r.p.m. Libraries were then centrifuged and resuspended in 1 ml SDCAA, serially diluted (10−1 to 10−5) in 250 μl deionized water, plated on tryptophan dropout plates and incubated for 2 days at 30 °C. The remaining library was expanded in 200 ml SDCAA for 2 days at 30 °C and 225 r.p.m. Colony counts from dropout plates were used to calculate library diversity. The library itself was stored at 4 °C until use.
Magnetic-activated cell sorting (MACS)
Constructed libraries were initially grown in SDCAA medium for 24 h at 30 °C, then transferred to SDGCAA medium at a starting OD 600 of 0.5–1.0 and incubated for 48 h at 20 °C or 24 h at 30 °C. After cultivation, yeast cells were collected, with 109 cells collected for the first round and 108 cells for subsequent rounds of sorting. Cells were pelleted at 2,500 g for 5 min, washed twice with PBSB and incubated with 300 nM biotinylated mouse or human CD98hc diluted in PBSB containing 1% milk for 3 h at room temperature with gentle mixing. Cells were then washed with ice-cold PBSB and pelleted at 2,500 g for 5 min. Streptavidin microbeads (Miltenyi Biotec, 130-048-101) were prepared in cold PBSB and incubated with cells for 40 min at 4 °C with steady mixing. Cells were washed again with cold PBSB, pelleted at 2,500 g for 5 min and kept on ice. LS columns (Miltenyi Biotec, 130-042-401) were mounted in two MACS magnets (Invitrogen, 12321) in series and primed with 5 ml cold PBSB. Cells were resuspended in 5 ml cold PBSB, passed through a 70-µm filter (Miltenyi Biotec, 130-098-462), loaded onto the MACS columns and washed with 5 ml cold PBSB. Afterwards, the columns were removed and filled with 5 ml SDCAA medium. Cells were eluted with the column plungers into 40 ml SDCAA. To evaluate library diversity, 200 µl of 10× and 100× dilutions of the elute were plated on tryptophan dropout plates. The remaining eluted library was supplemented with antibiotics as described above and cultured at 30 °C for 48 h.
Fluorescence-activated cell sorting (FACS)
The library was regrown and cultured as described in the MACS section. After induction, 107 cells per sorting condition were collected, washed three times with 1 ml of PBSB and pelleted at 16,000 g for 1 min. Cells were incubated for 3 h at room temperature with biotinylated mouse CD98hc and/or FLAG-tagged human CD98hc ectodomains at equivalent concentrations in PBSB containing 1% milk. CD98hc concentration was adjusted according to previous sorting rounds, and incubation volume was set to maintain an approximate 10:1 molar ratio of CD98hc to scFv. For samples containing FLAG-tagged human CD98hc ectodomains, a 1:200 dilution of rabbit anti-FLAG-tag antibody (Invitrogen, 701629) was added after 2 h for the final 1 h incubation. Cells were washed with ice-cold PBSB, pelleted and incubated on ice with mouse anti-myc tag antibody (Cell Signaling Technology, 2276S; 1:1,000 dilution) in PBSB for 15 min. Cells were washed again and then incubated in 1 ml PBSB with streptavidin Alexa Fluor 647 (Invitrogen, S32357; 1:1,000 dilution) and anti-mouse Fc antibody conjugated to Alexa Fluor 488 (Invitrogen, A11001; 1:200 dilution). For samples involving FLAG-tagged human CD98hc ectodomains, anti-rabbit Fc antibody conjugated to Alexa Fluor 568 (Invitrogen, A11011; 1:200) was also included. Following incubation, cells were washed again, pelleted and sorted for binding to mouse and/or human CD98hc using a Sony MA900 cell sorter. Sorted cells were collected and cultured as described in the MACS section.
Analysis of single clones from enriched libraries
Individual antibody sequences from enriched libraries were selected, constructed and cultured as described in the histidine scanning section. Binding to human and mouse CD98hc was assessed via flow cytometry at 300 nM CD98hc. Binding signal of each mutant to CD98hc was normalized by WT antibody binding to human CD98hc. Mutants with greater than 4% binding to both mouse and human CD98hc were classified as species-cross-reactive antibodies (such as MH17, MH24, D3), and others as non-cross-reactive. This dataset was used to assess the ability of four metrics: frequency, enrichment ratio, PSSM score and PSERM score, to predict antibody cross-reactivity.
NGS of libraries and data analysis of NGS datasets
Plasmid DNA of yeast libraries were extracted using the ZymoPrep Yeast Plasmid Miniprep kit (Zymo, D2004). Sequencing primers with Illumina adapters (p5 and p7) and indices (i5 and i7) for sample demultiplexing were designed69 and modified to include phasing elements43,70. The HCDR3 region of the scFv library was amplified using these primers and Q5 polymerase (New England Biolabs, M0491) in a two-round PCR process. PCR products were visualized on a 1% agarose gel, purified using the Qiagen PCR Gel Extraction kit (Qiagen, 28704) and quantified with the Qubit 1× dsDNA High Sensitivity Assay (Invitrogen, Q22321). Purified libraries were pooled and submitted to the University of Michigan Advanced Genomics Core for sequencing using the Illumina MiSeq technology with 2 × 300-bp paired-end reads.
Data were processed as described in previous work43. Paired-end fastq reads were merged with FLASH71, converted to fasta files and translated into protein sequences with Biopython72. Residues that were mutated during the library generation process were determined for each sequence and analysed with four different metrics (frequency, enrichment ratio, PSSM and PSERM), as described previously43,73.
Assessment of antibody affinities in scFv format using flow cytometry
Individual antibody sequences selected from the enriched libraries were constructed and expressed on yeast as described in the histidine scanning section. Affinities of the mutant antibodies for human, cyno and mouse CD98hc were evaluated by flow cytometry using serial dilutions of CD98hc proteins, as described in the histidine scanning section. The resulting data were processed using GraphPad Prism to determine the affinity of each antibody mutant.
Humanness and T-cell epitope predictions
The sequences of clinical-stage antibodies were obtained from the Therapeutic Antibody Database. Humanness scores were evaluated by AbNatiV46 in FASTA format, with the average of the AbNatiV V H and V L scores considered as the humanness score for each antibody. T-cell epitope prediction was performed on NetMHCIIpan-4.3 (ref. 47), with sequences input in FASTA format and peptide length set to 15 amino acids. Predictions were made against 27 major histocompatibility complex (MHC) molecules covering HLA DR, DQ and DP specificities (Supplementary Table 8)74. Resulting peptides in the top 2% rank were considered strong binders, and those between strong and weak thresholds (10%) were considered weak binders. Among all strong and weak binders, 15-mer peptides with identical predicted core and MHC were grouped, retaining only the strongest binders by % rank. The germlines of each evaluated antibody obtained from ANARCI75 and IMGT76 databases were analysed using the same process for T-cell epitope predictions. Prediction results for each antibody were compared to germlines to identify whether peptide cores were germline encoded. The number of predicted T-cell epitopes per antibody was calculated by excluding germline sequences from the total unique strong-binding core peptides.
Computational epitope and paratope analysis of CD98hc antibodies
Structural paratopes of antibodies and other engineered protein binders, along with the corresponding epitopes of human CD98hc and buried surface areas, were derived from the co-crystal structures of human CD98hc/binder complexes in the Protein Data Bank (Supplementary Table 14). In this context, the epitope is defined as amino acids in human CD98hc within 4 Å of any binder atom, and the paratope as amino acids in the binder within 4 Å of any atom in human CD98hc. Calculations were performed using Afpdb77. Solvent-accessible surface area (SASA) of the WT Fv fragment was calculated in unbound form (SASA_free) and in complex with human CD98hc (SASA_complex; PDB ID 7DF1). Buried surface area (BSA), defined as SASA_free−SASA_complex, represents the surface area shielded from solvent upon antigen binding. Relative buried surface area (rBSA) was calculated as BSA/SASA_free. Total and mean BSA values were calculated for each CDR loop to assess their overall contribution to the interaction. All solvent accessibility calculations were performed using the Shrake–Rupley algorithm implemented in Biopython72.
Antibody production
To produce antibodies, 15 µg of total DNA, consisting of 75% 10 mg ml−1 single-stranded DNA (Sigma-Aldrich, D7656) and 25% plasmid DNA, was prepared for transfection. Plasmid DNA consisted of equal mass ratios of all 2 or 3 chains for IgGs and bispecific antibodies, respectively. Transfection and cell collection was performed as described in the antigen expression section.
For antibody purification, supernatants were incubated overnight with Protein A agarose beads (Thermo Fisher, PI20334) at 4 °C with shaking. Beads were collected in centrifuge columns (Thermo Fisher, PI89898), washed 3 times with 10 ml of PBS and eluted with 0.1 M glycine buffer (pH 3.0) after 15 min incubation. Eluted antibodies were neutralized and exchanged into 20 mM sodium acetate buffer (pH 5.0) using desalting columns (Thermo Fisher, 89891) and stored at −80 °C. Antibody characterization was performed as described in the antigen expression section.
Surface plasmon resonance (SPR)
The affinities of four antibodies (WT, D4-4, 3.5D19 and 3.5D5) for mouse, human and cyno CD98hc were determined using SPR on a Nicoya Alto instrument (v.1.2, Nicoya). IgGs were immobilized on 16-channel CMD Carboxyl Cartridges (Nicoya, KC-CBX-CMD-16) via the Carboxyl Surfacing kit (Nicoya, ALTO-R-CBX-SURF, all reagents used in the subsequent steps were included in this kit). The multicycle kinetics mode with five cycles was used, each including specific association, dissociation and surface regeneration in accordance with Nicoya’s standard protocols. Protein G (50 µg ml−1 in 10 mM sodium acetate, pH 4.5) was covalently linked to 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide (EDC/NHS) (200 mM)-activated sensors for 10 min, followed by blocking of unreacted groups with 1 M ethanolamine (pH 8.5) for 5 min. Antibodies (25 nM in 0.05% PBST) were immobilized via Protein G for 5 min. CD98hc antigens were prepared in the same buffer at threefold serial dilutions: 500–6.2 nM for mouse, and 333.3–4.1 nM for human and cyno. Binding measurements were performed at 25 °C, with association and dissociation phases of 6- and 20-min. Regeneration was carried out using 10 mM glycine-HCl (pH 2.0) for 1 min, followed by reloading with 25 nM IgG for the next cycle. Data were analysed using Nicosystem (Nicoya) with a 1:1 Langmuir binding model. Each interaction was measured 2–4 times.
Enzyme-linked immunosorbent assay (ELISA)
Binding affinity and epitope of bispecific antibodies were determined by ELISA. For affinity assessment, 96-well plates were coated with recombinant mouse, human or cyno CD98hc ectodomains at 10 µg ml−1 overnight at 4 °C with shaking. For epitope mapping, mouse, human and cyno CD98hc proteins were engineered with glycine-serine linkers replacing the predicted antibody-binding regions (loop 1 and loop 2 mutants). Wild-type and mutant antigens were coated onto ELISA plates at 10 µg ml−1 under the same conditions. Plates were washed three times with PBST (0.5% Tween-20 in 1× PBS) and blocked with 3% BSA (Sigma-Aldrich, A7906) in PBS for 1 h at room temperature. After washing, a dilution series of bispecific antibodies in PBS with 1% BSA was added and incubated for 2 h at room temperature. For epitope detection, an anti-6×His tag monoclonal antibody (Thermo Fisher, 4E3D10H2/E3) was used to bind the His-tagged antigens. Plates were washed three times with PBST. Anti-human Fc HRP-conjugated secondary antibody (Thermo Fisher, A18817) diluted 1:40,000 in PBS was added for bispecific antibody detection. Anti-mouse Fc HRP-conjugated secondary antibody (Jackson ImmunoLabs, 115-035-164) diluted 1:30,000 in PBS was used for His-tagged antigen detection. Plates were incubated for 1 h at room temperature, washed, and 100 µl TMB substrate (Thermo Fisher, ENN301) was added per well. After 5 min in the dark, the reaction was stopped with 100 µl 0.18 M sulfuric acid (Thermo Fisher, A300-212). Absorbance at 450 nm was recorded using a plate reader (Molecular Devices Spectramax). Results were analysed with GraphPad Prism.
Experimental epitope and functional paratope analysis of CD98hc antibodies
The epitope of D4-4 was determined through alanine scan of loops 1 and 2 on mouse and human CD98hc ectodomains. Individual residues in either loop 1 or loop 2 were mutated to alanine (or glycine if WT was alanine). Variants were expressed as recombinant proteins and purified to >85% monomer. Binding to IgG/D4-4 was tested via ELISAs using an antibody concentration of 50 nM.
The functional paratope of D4-4 was determined using alanine and reversion scans. Alanine mutants of D4-4 were generated by individually substituting all residues that differ between WT and D4-4 with alanine (or glycine for residues already encoded as alanine). Reversion mutants were generated by mutating residues altered from WT to D4-4 back to the original WT residue. All variants were expressed on yeast. Binding of the variants to human and mouse CD98hc was assessed via flow cytometry at a CD98hc concentration of 100 nM. The binding signal of each mutant was normalized to WT antibody binding and analysed on GraphPad Prism software.
Melting temperature analysis
The melting temperature (T m ) of bispecific antibodies was evaluated using differential scanning fluorimetry. Antibodies were diluted to 0.12 mg ml−1 in 20 mM sodium acetate buffer (pH 5.0) and mixed with Protein Thermal Shift Dye (Applied Biosystems, 4461146) at a 1× final concentration, with buffer-only serving as blanks. Thermal scanning was performed at the University of Michigan Advanced Genomics Core using a QuantStudio Real-Time PCR System. Temperature was ramped from 25 to 98 °C with triplicate measurements. Data analysis was performed using GraphPad Prism software.
Non-specific binding analysis
The soluble membrane protein (SMP) fraction was prepared according to established methods78 and was biotinylated using the same protocol described for CD98hc ectodomains. The non-specific binding (polyspecificity particle79) assay was performed as follows. Protein A magnetic beads (Invitrogen, 10002D) were washed and diluted to 54 μg ml−1 in PBSB. A 30-μl aliquot of beads was incubated overnight with 85 μl of antibody (15 μg ml−1). Beads were then washed twice via centrifugation (3,500 g for 4 min) and resuspended in PBSB. Subsequently, beads were incubated with 0.1 mg ml−1 of biotinylated SMP for 20 min at 4 °C. Following a wash step, a 1:1,000 dilution of streptavidin Alexa Fluor 647 (Invitrogen, S32357) was applied and incubated on ice for 4 min. After a final wash and resuspension in PBSB, samples were analysed by flow cytometry for median fluorescence intensity (MFI). The polyspecificity score for each IgG was derived from 9 independent replicates and calculated as: (target IgG MFI−elotuzumab MFI)/(emibetuzumab MFI−elotuzumab MFI).
Antibody self-association analysis
Antibody self-association was measured using charge-stabilized self-interaction nanoparticle spectroscopy, as previously described80. Gold nanoparticles (Ted Pella, 157051) were coated with anti-human capture antibody (Jackson ImmunoResearch, 109-005-008) and polylysine (Merck, P1274) at a 90:10 (w/w) ratio overnight. Gold conjugates (5 μl) were mixed with 45 μl of dilute antibody (11.1 μg ml−1) and incubated for 4 h at room temperature. Absorbance spectra (450–650 nm, 1 nm increments) were acquired using a Biotek Synergy Neo plate reader (Agilent BioTek). The plasmon wavelength was determined by the maximum absorbance and normalized using a calibration panel of antibodies (NIST, ibalizumab, mepolizumab, trastuzumab and romosozumab), thereby rescaling the measurements to the established metric reported80.
Pharmacokinetic analysis
Radiolabelling of antibodies was performed using [125I] NaI (Perkin Elmer) and the Pierce iodination reagent (Thermo Fisher, 28601). Labelled antibodies were purified using Zeba desalting columns, and radiochemical integrity was evaluated by thin-layer chromatography on aluminum silica gel plates (Millipore Sigma, 105554) with 75% methanol and 25% 1 M sodium acetate (pH 6.8). Radiolabelling efficiency exceeded 75%, and only samples with <5% free [125I] were used. For radiotracing studies, ~1 μg of [125I]-labelled antibody was combined with unlabelled antibody to reach a target radioactivity of 1 × 106 counts per minute, as confirmed using a Wizard2 2470 Gamma counter (Perkin Elmer, 2470-0050).
For pharmacokinetic analysis in WT mice (C57BL/6J male mice, aged 8–16 weeks; Jackson Laboratory, 000664) and transgenic human CD98hc mice (C57BL/6N-Slc3a2tm1(SLC3A2)Bcgen/Bcgen female mice, aged 8–16 weeks; Biocytogen, 110983), doses were standardized to either 20 or 60 nmol kg−1, formulated in sterile PBS and filtered before administration. Mice were anaesthetized with isoflurane (Henry Schein, 66794-017-25), and doses were administered intravenously via the retroorbital plexus. Terminal pharmacokinetic data were collected at 1 h, 1 day, 7 days or 7.5 days post administration (n = 4 per timepoint). At terminal timepoints, mice were anaesthetized, blood was collected, and mice were perfused with PBS. Organs were collected, weighed and radioactivity quantified by gamma counter. Data were analysed using GraphPad Prism. All mice were maintained on a 12-h light/dark cycle under controlled environmental conditions, with an ambient temperature of 22 °C and 30–70% relative humidity.
Evaluation of CD98hc levels on mouse brain endothelial cells
Mouse brains (WT and humanized CD98hc) were collected after transcardial perfusion with ice-cold PBS to remove circulating blood. Brain tissue was placed in ice-cold PBS and processed using the Miltenyi Biotec Adult Brain Dissociation kit (130-107-677). Cells were stained with anti-mouse CD45-BV421 (BioLegend, 103133), anti-mouse CD31-Alexa Fluor 488 (R&D Systems, FAB3628G) and anti-CD98hc (WT S1-F4 antibody for humanized mice and CP1 for WT mice, Alexa Fluor 647-labelled) antibodies for 1 h at 4 °C in the dark. Samples were washed twice in flow buffer (PBSB) and resuspended. For live/dead discrimination, 7-AAD (BioLegend, 420404) was added immediately before acquisition. Samples were acquired on a Bio-Rad ZE5 Cell Analyzer. Cells were gated to exclude debris and doublets, and live cells were identified as 7-AAD negative. Endothelial cells were defined as CD31+, and within this population, MFI of Alexa Fluor 647-labelled marker was quantified. For receptor density, calibration beads (Bangs Laboratories, Quantum Simply Cellular, 816A) were incubated with the same Alexa Fluor 647-conjugated antibody. Beads were prepared in parallel and acquired under identical settings. A calibration curve relating MFI to antibody binding capacity was generated, allowing conversion of sample MFI to receptor numbers per cell. MFI was multiplied by 2 to account for bivalent IgG binding.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.





© All Rights Reserved.